






















Inês Mendes
Bioinformatics PhD student.
Labmeeting
8 April 2022
Inês Mendes
MRamirez Lab - iMM
@ines_cim
cimendes
metagenomic de novo assembly
finding the best fit in a world of options
Bacterial Population Genetics
Pathogenesis and Natural History of Infection
Outbreak Investigation and Control
Surveillance of Infectious Diseases
Microbial Genomics
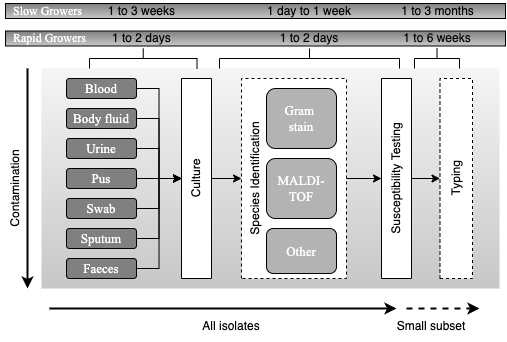
| The now
| The now
Joana Silva & Ana Friães
| The now
| The now
| The now
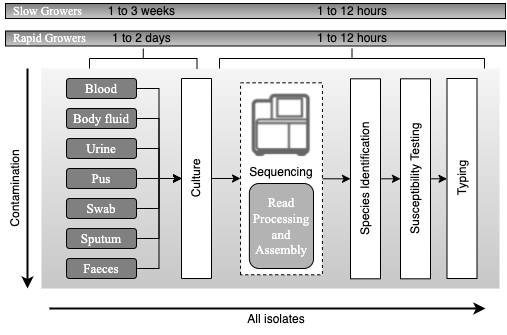
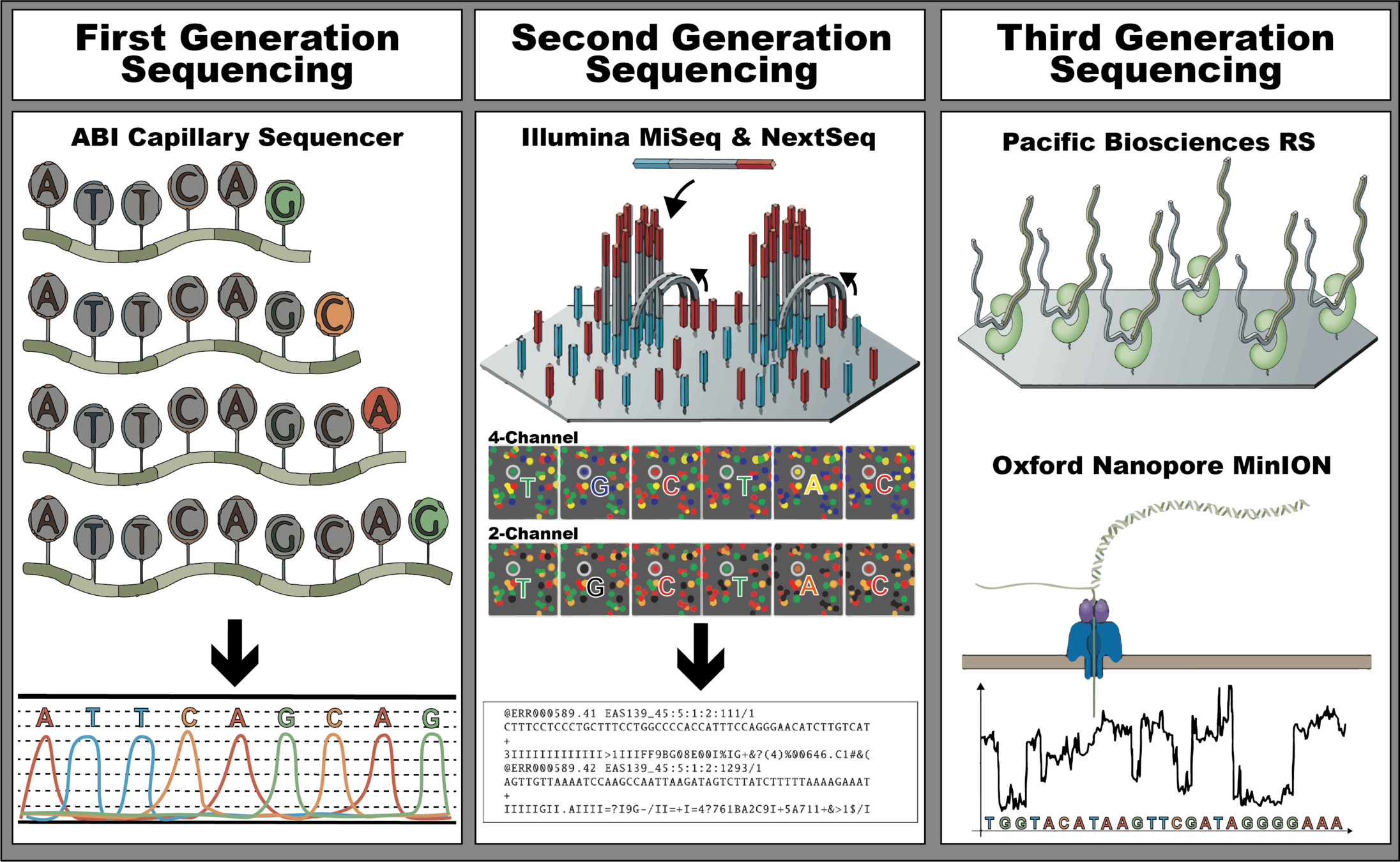
| The future
| The future
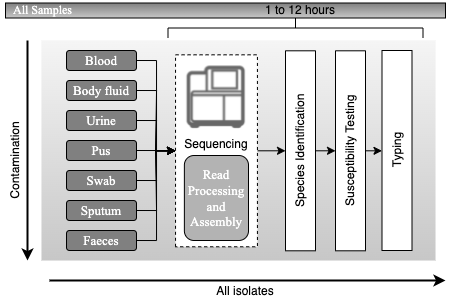
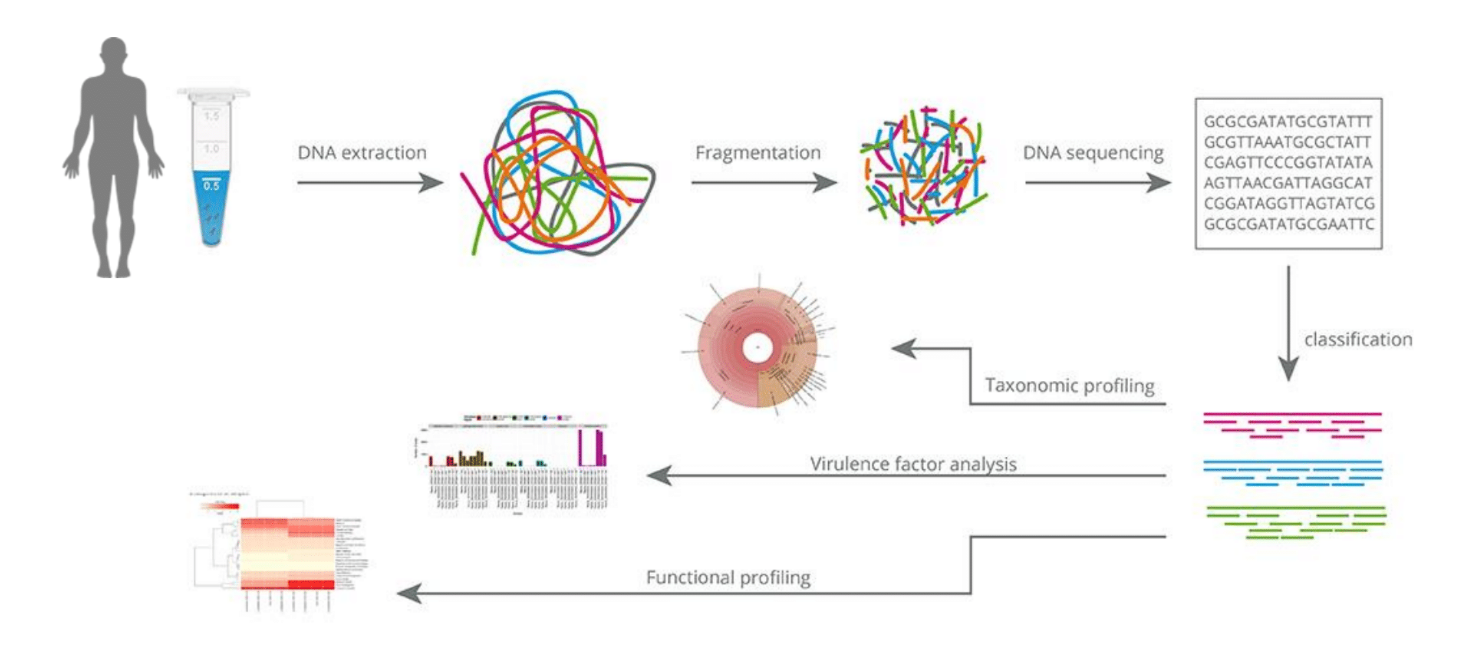
Metagenomics
Random "shotgun" sequencing of microbial DNA, without selecting a particular gene or species.
| Assembly
The assembly methods provide longer sequences that are more informative than shorter sequencing data and can provide a more complete picture of the microbial community in a given sample.
Reads
Contigs
Genomes
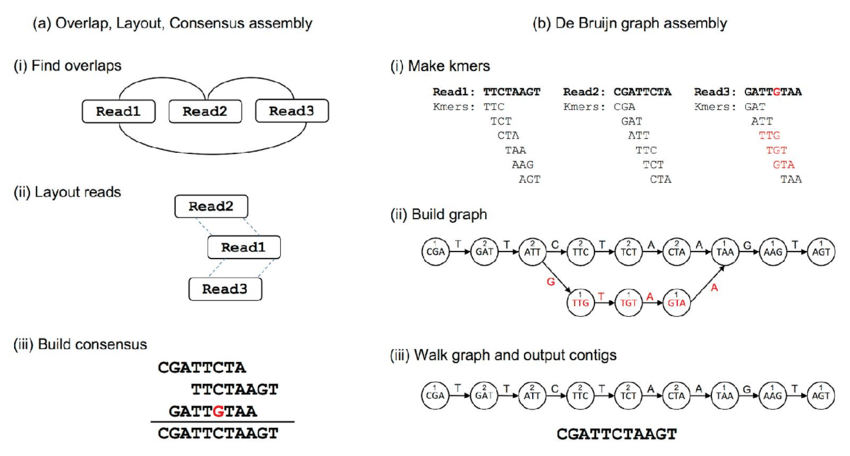
| de novo Assembly
Martin Ayling, Matthew D Clark, Richard M Leggett, New approaches for metagenome assembly with short reads, Briefings in Bioinformatics, Volume 21, Issue 2, March 2020, Pages 584–594, https://doi.org/10.1093/bib/bbz020
| de novo Assembly
Major issues
Reads
Contigs
Genomes
https://github.com/B-UMMI/LMAS
https://lmas.readthedocs.io/
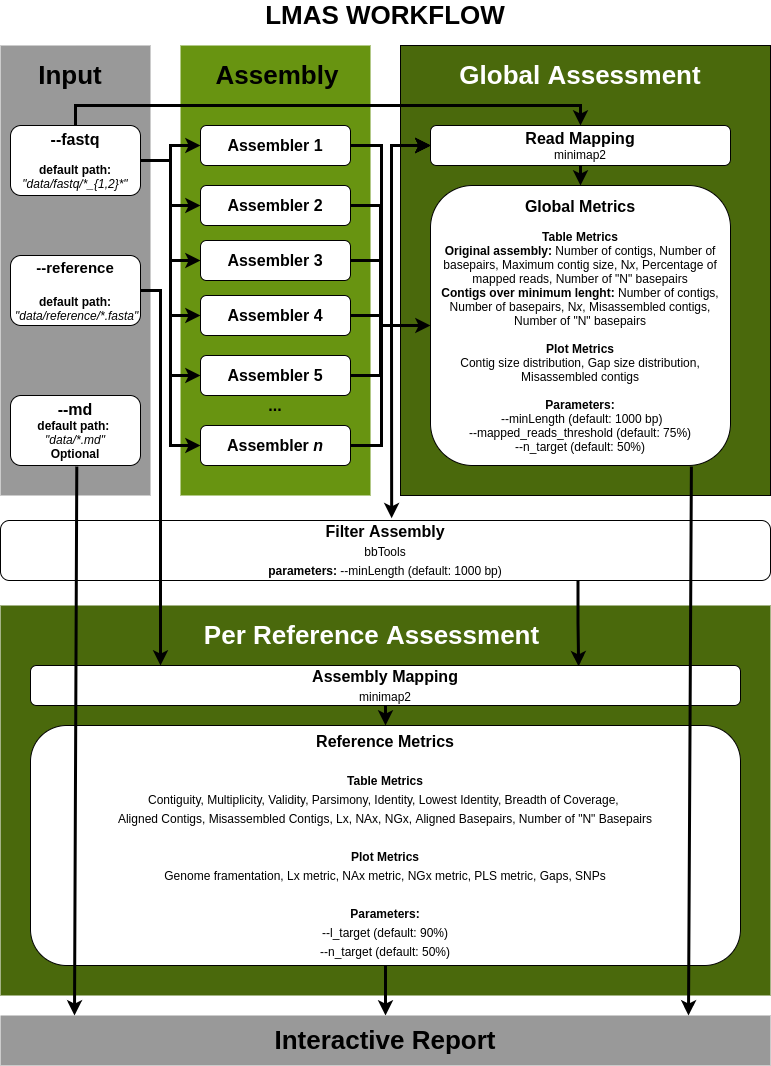
| Last Metagenomic Assembler Standing
Automated workflow enabling the benchmarking of genomic and metagenomic prokaryotic de novo assembly software using defined mock communities.
| Last Metagenomic Assembler Standing
A container engine (Docker, singularity, shifter...).
apt-install docker-ceInstall LMAS
conda install -c bioconda LMASRun LMAS
LMAS --fastq <reads_{1,2}.fq.gz> --reference <reference.fasta>| Last Metagenomic Assembler Standing
The input data is assembled in parallel by the set of genomic and metagenomic de novo assemblers in LMAS.
The global and per reference metrics are grouped in the interactive LMAS report for exploration.
The resulting assembled sequences are processed and assembly quality metrics are computed.
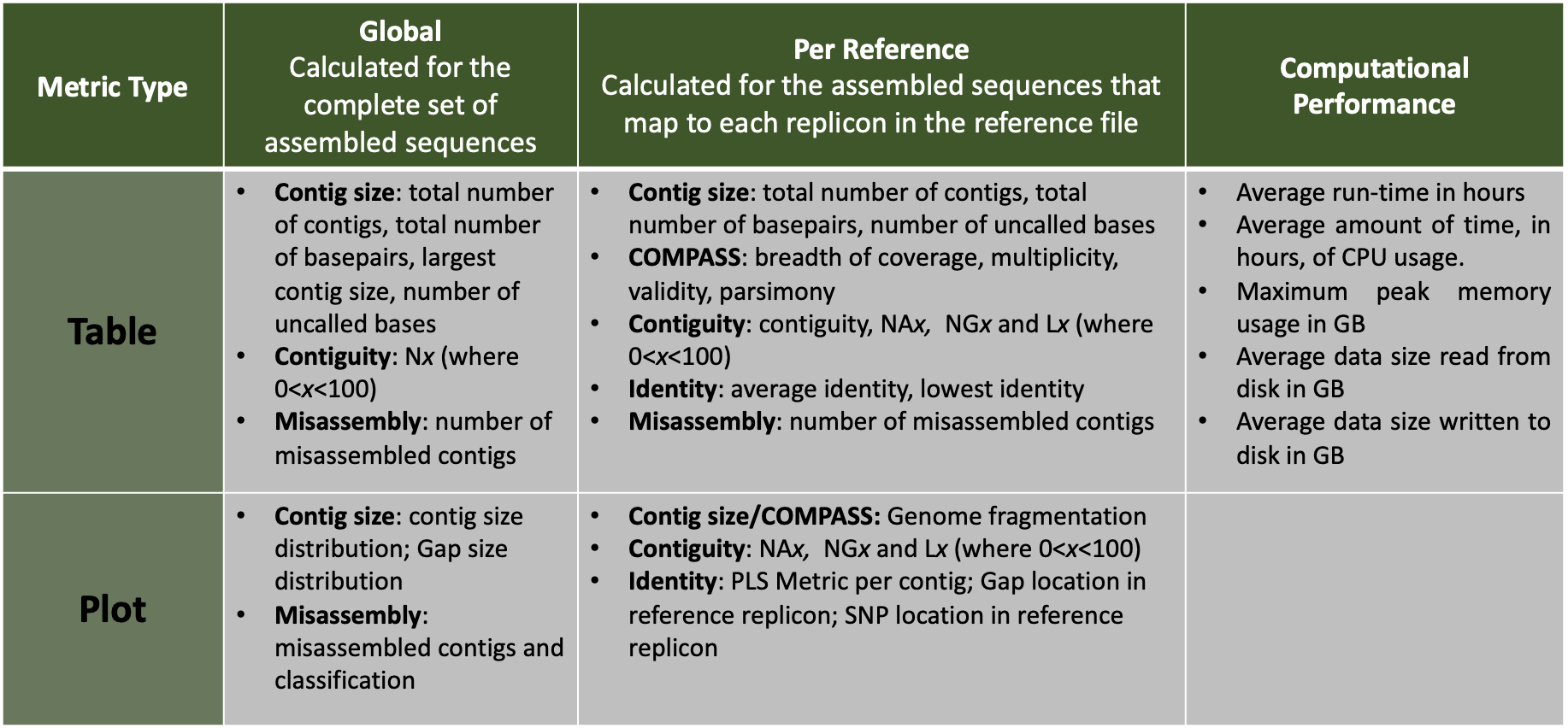
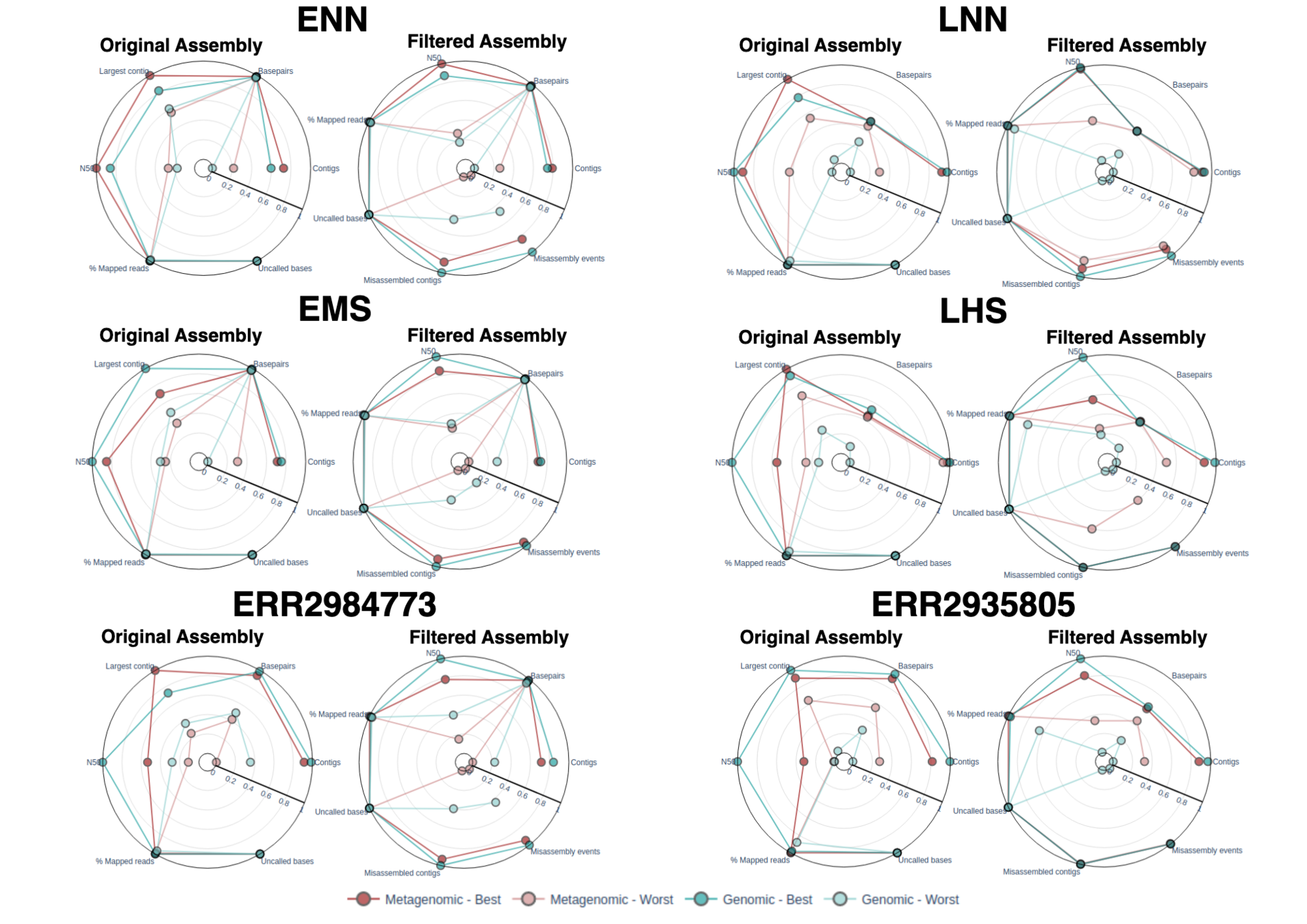
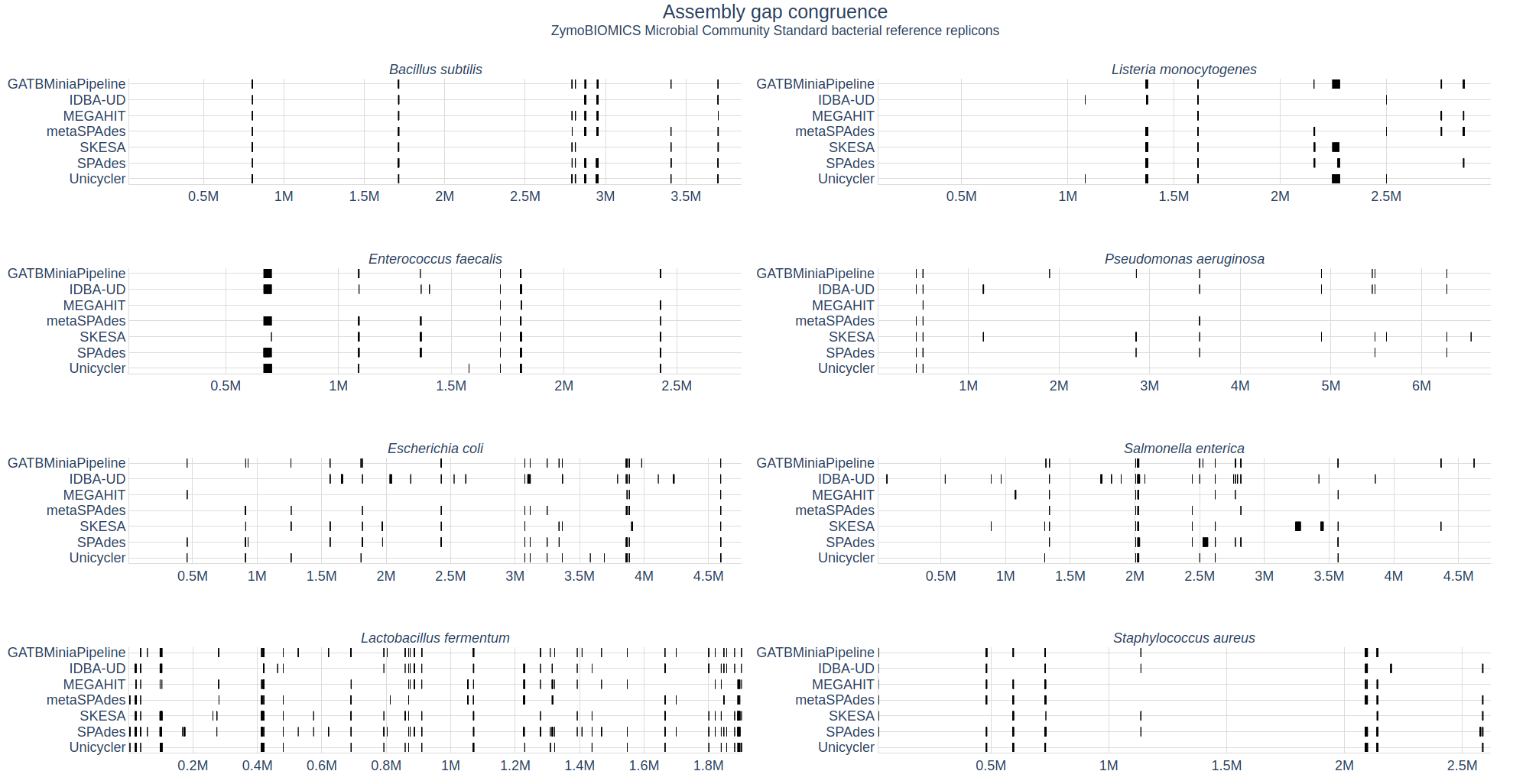
| Assembly Quality Metrics
The tabular presentation allows direct comparison of exact values between assemblies, and the interactive plots allow for an intuitive overview and easy exploration of results.
| Sample | Distribution | Error Model | Read Pairs (M) |
|---|---|---|---|
| ENN | Even | None | 8.6 |
| EHS | Even | Illumina HiSeq | 8.6 |
| ERR2984773 | Even | Real Sample | 8.6 |
| LNN | Log | None | 47.5 |
| LHS | Log | Illumina HiSeq | 47.5 |
| ERR2935805 | Log | Real Sample | 47.5 |
| ZymoBIOMICS Microbial Community Standards
hearding the LMAS
Evaluating metagenomic long and short de novo assembly methods
LMAS --wf ont \
--fastq <reads_{1,2}.fq.gz> \
--reference <reference.fasta>LMAS --wf hybrid \
--fastq <reads_{1,2}.fq.gz> \
--reference <reference.fasta>| LMAS for long noisy ONT reads
Automated workflow enabling the benchmarking of long-read genomic and metagenomic prokaryotic de novo assembly software using defined mock communities.
| LMAS for long and short reads
Automated workflow enabling the benchmarking of short and long-read genomic and metagenomic prokaryotic hybrid de novo assembly software using defined mock communities.
Special thanks to Pedro Vila-Cerqueira, Rafael Mamede and Mário Ramirez.
FCT PhD Grants SFRH/BD/129483/2017
COVID/BD/152618/2022
MRamirez Lab, iMM
2019
By Inês Mendes
Lab meeting 8 April 2022




