





























Inês Mendes
Bioinformatics PhD student.
GIP Research Meeting
@ines_cim
cimendes
Inês Mendes
The needs:
Runs the same regardless of the environment.
Enables the distribution and deployment of scientific software in a runnable state.
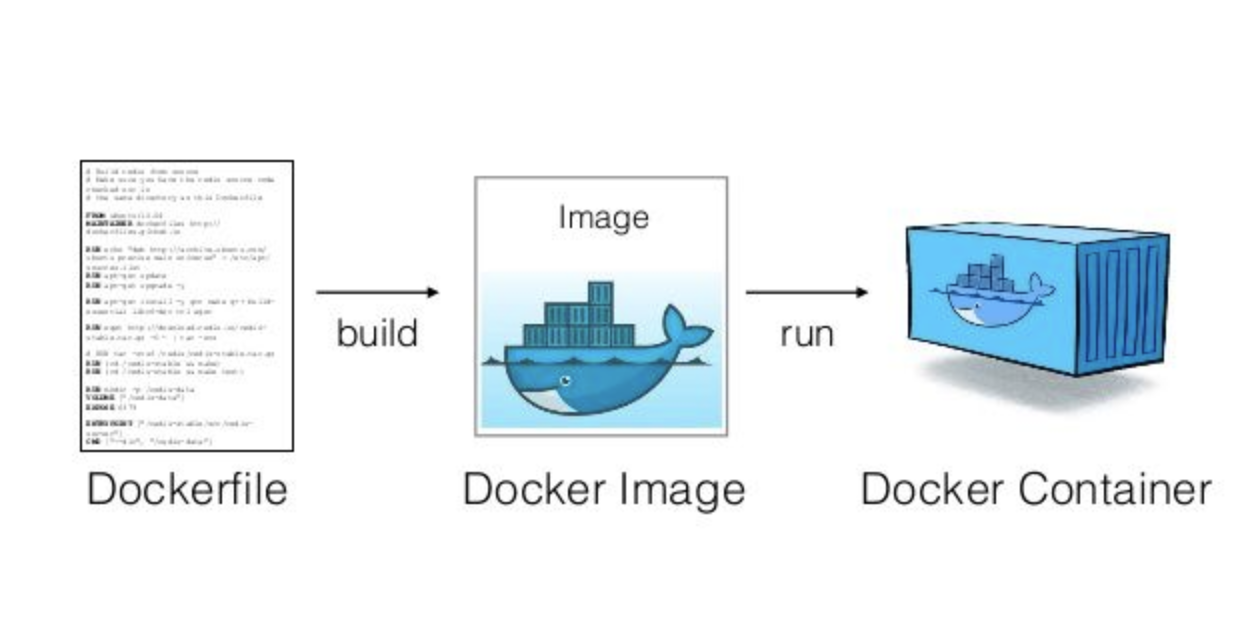
A container image is a lightweight, stand-alone, executable package of a software that includes everything needed to run it:
Host Hardware
Host Hardware
Container Engine
Host OS
Host OS
Hypervisor
Guest OS
Guest OS
App
App
Guest OS
VM1
VM2
App
App
App
App
Virtual Machines
Containers
build
pull & run
host
push
Enables scalable and reproducible scientific workflows using software containers. It simplifies the deployment of complex parallel and reactive workflows.
Reactive workflow framework
Create pipelines with asynchronous (and implicitly parallelized) data streams
Programing DSL
Has its own language for building a pipeline
Containerized
Out of the box integration with containers engines (Docker, Singularity, Shifter)
Installation
Ubiquitous on UNIX. Windows: Cygwin or Linux sysbsystem, maybe...sudo apt-get install openjdk-8-jdkcurl -s https://get.nextflow.io | bashOptional (but recommended)
The creation of Nextflow pipelines was designed for bioinformaticians familiar with programming.
It's execution is for everyone.
The game changing combination of nextflow + containers:
Substantial challenges still persist:
Workflow based development
Component based development
Components are modular pieces of nextflow code with some basic rules:
Component A
- Input/Output
- Parameters
- Resources
Component B
- Input/Output
- Parameters
- Resources
With this framework, building workflows becomes simple:
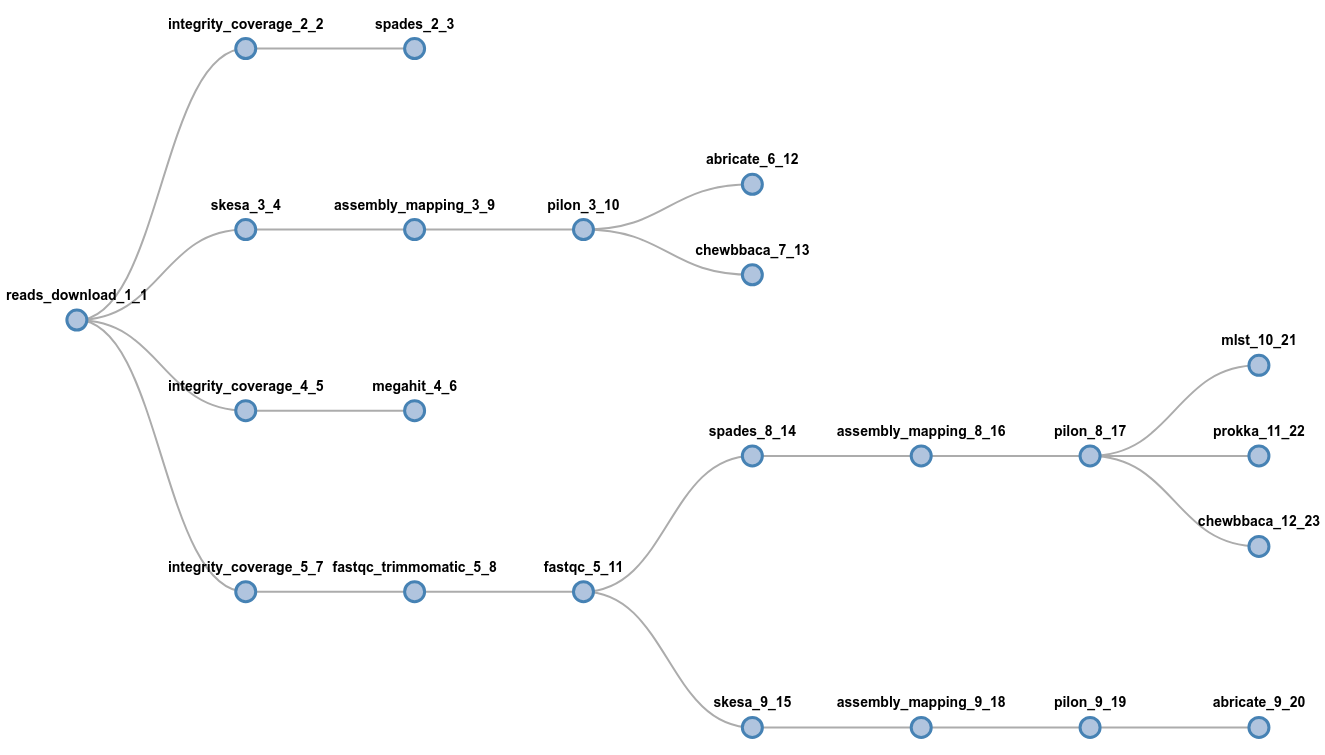
flowcraft build -t 'trimmomatic fastqc spades pilon' -o my_nextflow_pipelineResults in the following workflow DAG
$ nextflow run my_nextflow_pipeline.nf --help
N E X T F L O W ~ version 0.32.0
Launching `my_nextflow_pipeline.nf` [jovial_swirles] - revision: b4473f5a12
============================================================
F L O W C R A F T
============================================================
Built using flowcraft v1.4.0
Usage:
nextflow run my_nextflow_pipeline.nf
--fastq Path expression to paired-end fastq files. (default: fastq/*_{1,2}.*) (default: 'fastq/*_{1,2}.*')
Component 'INTEGRITY_COVERAGE_1_1'
----------------------------------
--genomeSize_1_1 Genome size estimate for the samples in Mb. It is used to estimate the coverage and other assembly parameters andchecks (default: 1)
--minCoverage_1_1 Minimum coverage for a sample to proceed. By default it's setto 0 to allow any coverage (default: 0)
Component 'TRIMMOMATIC_1_2'
---------------------------
--adapters_1_2 Path to adapters files, if any. (default: 'None')
--trimSlidingWindow_1_2 Perform sliding window trimming, cutting once the average quality within the window falls below a threshold (default: '5:20')
--trimLeading_1_2 Cut bases off the start of a read, if below a threshold quality (default: 3)
--trimTrailing_1_2 Cut bases of the end of a read, if below a threshold quality (default: 3)
--trimMinLength_1_2 Drop the read if it is below a specified length (default: 55)
--clearInput_1_2 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)
Component 'FASTQC_1_3'
----------------------
--adapters_1_3 Path to adapters files, if any. (default: 'None')
Component 'SPADES_1_4'
----------------------
--spadesMinCoverage_1_4 The minimum number of reads to consider an edge in the de Bruijn graph during the assembly (default: 2)
--spadesMinKmerCoverage_1_4 Minimum contigs K-mer coverage. After assembly only keep contigs with reported k-mer coverage equal or above this value (default: 2)
--spadesKmers_1_4 If 'auto' the SPAdes k-mer lengths will be determined from the maximum read length of each assembly. If 'default', SPAdes will use the default k-mer lengths. (default: 'auto')
--clearInput_1_4 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)
--disableRR_1_4 disables repeat resolution stage of assembling. (default: false)
Component 'ASSEMBLY_MAPPING_1_5'
--------------------------------
--minAssemblyCoverage_1_5 In auto, the default minimum coverage for each assembled contig is 1/3 of the assembly mean coverage or 10x, if the mean coverage is below 10x (default: 'auto')
--AMaxContigs_1_5 A warning is issued if the number of contigs is overthis threshold. (default: 100)
--genomeSize_1_5 Genome size estimate for the samples. It is used to check the ratio of contig number per genome MB (default: 2.1)
Component 'PILON_1_6'
---------------------
--clearInput_1_6 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)Help and parameters tailor-made to the pipeline
It's easy to get experimental:
flowcraft build -t 'trimmomatic fastqc skesa pilon' -o my_nextflow_pipelineSwitch spades for skesa
flowcraft build -t 'trimmomatic fastqc skesa pilon (abricate | prokka)' -o my_nextflow_pipelineAdd genome annotation components in the end
It's easy to get wild:
flowcraft build -t 'reads_download (
spades | skesa pilon (abricate | chewbbaca) | megahit |
fastqc_trimmomatic fastqc (spades pilon (
mlst | prokka | chewbbaca) | skesa pilon abricate))'
-o my_nextflow_pipelinewait, what?
Forks
Connect one component to multiple
Secondary channels
Connect non-adjacent components
Extra inputs
Inject user input data anywhere
Recipes
Curated and pre-assembled pipelines for specific needs
Tracks Nextflow execution in real time:
Dynamic generation of interactive report page
Multiple Raw Input Types
Not limited to paired-end FastQ or Fasta
Dynamic Input in Components
One component, multiple inputs
Expand Building Features
New merge operators
Diogo N Silva
Tiago F Jesus
Inês Mendes
Bruno
Ribeiro-Gonçalves
Prof. Mário Ramirez
Prof. João A Carriço
and happy pipeline building
conda install flowcraftpip install flowcraftFCT PhD Grant SFRH/BD/129483/2017
Funding and Acknowledgements
By Inês Mendes
GIP Research Meeting - 12/03/2019 UMCG



