





















Inês Mendes
Bioinformatics PhD student.
Applied Bioinformatics and Public Health Microbiology
@ines_cim
cimendes
Inês Mendes
05 - 07 June 2019
The game changing combination of nextflow + containers:
Substantial challenges still persist:
Workflow based development
Component based development
Components are modular pieces of nextflow code with some basic rules:
Component A
- Input/Output
- Parameters
- Resources
Component B
- Input/Output
- Parameters
- Resources
With this framework, building workflows becomes simple:
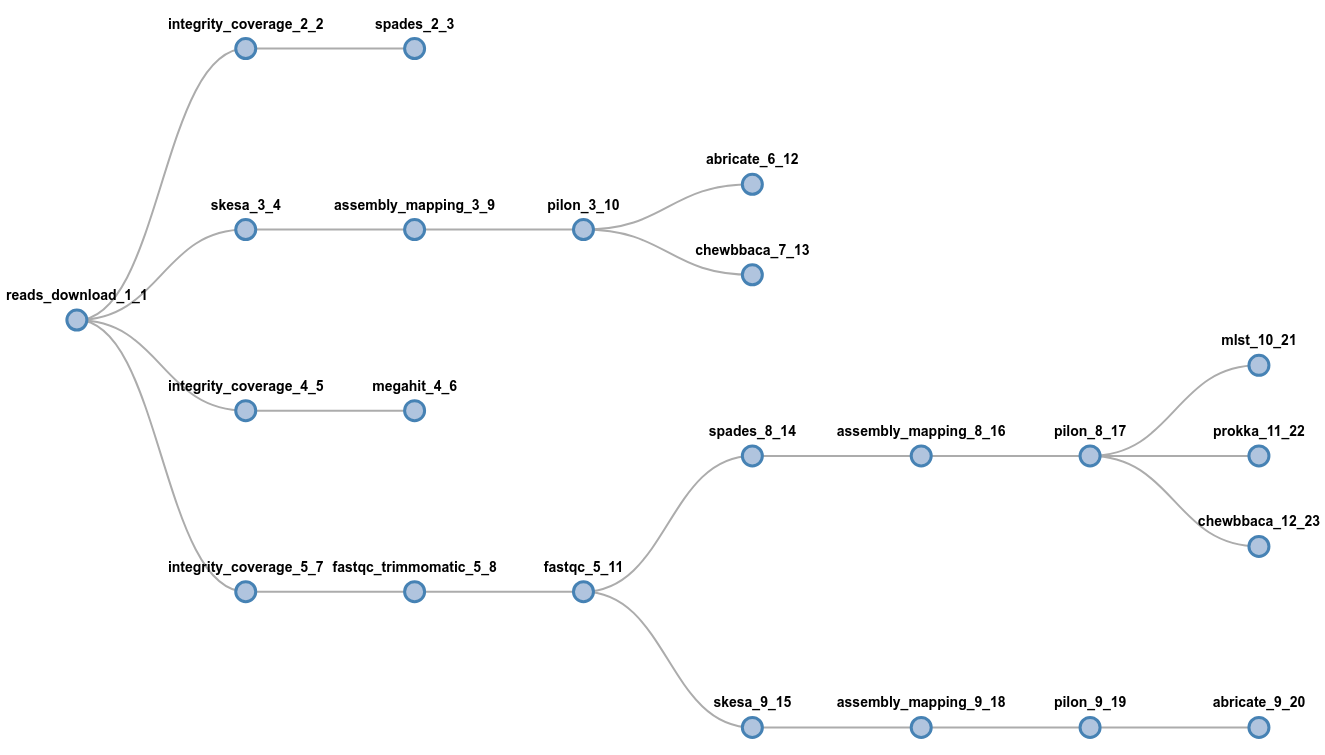
flowcraft build -t 'trimmomatic fastqc spades pilon' -o my_nextflow_pipelineResults in the following workflow DAG
$ nextflow run my_nextflow_pipeline.nf --help
N E X T F L O W ~ version 0.32.0
Launching `my_nextflow_pipeline.nf` [jovial_swirles] - revision: b4473f5a12
============================================================
F L O W C R A F T
============================================================
Built using flowcraft v1.4.0
Usage:
nextflow run my_nextflow_pipeline.nf
--fastq Path expression to paired-end fastq files. (default: fastq/*_{1,2}.*) (default: 'fastq/*_{1,2}.*')
Component 'INTEGRITY_COVERAGE_1_1'
----------------------------------
--genomeSize_1_1 Genome size estimate for the samples in Mb. It is used to estimate the coverage and other assembly parameters andchecks (default: 1)
--minCoverage_1_1 Minimum coverage for a sample to proceed. By default it's setto 0 to allow any coverage (default: 0)
Component 'TRIMMOMATIC_1_2'
---------------------------
--adapters_1_2 Path to adapters files, if any. (default: 'None')
--trimSlidingWindow_1_2 Perform sliding window trimming, cutting once the average quality within the window falls below a threshold (default: '5:20')
--trimLeading_1_2 Cut bases off the start of a read, if below a threshold quality (default: 3)
--trimTrailing_1_2 Cut bases of the end of a read, if below a threshold quality (default: 3)
--trimMinLength_1_2 Drop the read if it is below a specified length (default: 55)
--clearInput_1_2 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)
Component 'FASTQC_1_3'
----------------------
--adapters_1_3 Path to adapters files, if any. (default: 'None')
Component 'SPADES_1_4'
----------------------
--spadesMinCoverage_1_4 The minimum number of reads to consider an edge in the de Bruijn graph during the assembly (default: 2)
--spadesMinKmerCoverage_1_4 Minimum contigs K-mer coverage. After assembly only keep contigs with reported k-mer coverage equal or above this value (default: 2)
--spadesKmers_1_4 If 'auto' the SPAdes k-mer lengths will be determined from the maximum read length of each assembly. If 'default', SPAdes will use the default k-mer lengths. (default: 'auto')
--clearInput_1_4 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)
--disableRR_1_4 disables repeat resolution stage of assembling. (default: false)
Component 'ASSEMBLY_MAPPING_1_5'
--------------------------------
--minAssemblyCoverage_1_5 In auto, the default minimum coverage for each assembled contig is 1/3 of the assembly mean coverage or 10x, if the mean coverage is below 10x (default: 'auto')
--AMaxContigs_1_5 A warning is issued if the number of contigs is overthis threshold. (default: 100)
--genomeSize_1_5 Genome size estimate for the samples. It is used to check the ratio of contig number per genome MB (default: 2.1)
Component 'PILON_1_6'
---------------------
--clearInput_1_6 Permanently removes temporary input files. This option is only useful to remove temporary files in large workflows and prevents nextflow's resume functionality. Use with caution. (default: false)Help and parameters tailor-made to the pipeline
It's easy to get wild:
flowcraft build -t 'reads_download (
spades | skesa pilon (abricate | chewbbaca) | megahit |
fastqc_trimmomatic fastqc (spades pilon (
mlst | prokka | chewbbaca) | skesa pilon abricate))'
-o my_nextflow_pipelinewait, what?
Forks
Connect one component to multiple
Secondary channels
Connect non-adjacent components
Extra inputs
Inject user input data anywhere
Recipes
Curated and pre-assembled pipelines for specific needs
Multiple Raw Input Types
Not limited to paired-end FastQ or Fasta
Dynamic Input in Components
One component, multiple inputs
Expand Building Features
New merge operators
Diogo N Silva
Tiago F Jesus
Inês Mendes
Bruno
Ribeiro-Gonçalves
Prof. Mário Ramirez
Prof. João A Carriço
conda install flowcraftFCT PhD Grant SFRH/BD/129483/2017
BacGenTrack project [FCT / Scientific and Technological Research Council of Turkey, TUBITAK/0004/2014]
Funding and Acknowledgements
brew install brewsci/bio/flowcraftpip install flowcraftBy Inês Mendes
3rd meeting bioinformatics in medical microbiology NL - March 11th in Utrecht CS



