Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides are current as of August-23-2023
These slides at https://slides.com/jbloom/new_2nd_gen_ba2_variant
This is analysis of spike amino-acid mutations in the new SARS-CoV-2 variant BA.2.86. Although only a modest number of sequences have been detected so far, they come from multiple countries, indicating at least some global transmission. In addition, the variant is present in some wastewater samples (see here and here).
One possible scenario is that BA.2.86 is less transmissible than current dominant variants, and so never spreads widely. This is the fate of most new SARS-CoV-2 variants. However, another possible scenario is BA.2.86 has a sufficient combination of antigenic advantage and inherent transmissibility to spread widely, as has happened with other variants such as the original Omicron or XBB. There are currently not enough data to be confident about which of these scenarios will occur for BA.2.86.
Regardless, we can use deep mutational scanning to estimate how spike mutations affect antigenicity and ACE2 affinity. It is scientifically valuable to do this, even when we do not yet know how widely the variant will spread.
The new variant has been named BA.2.86, and appears to be descended from BA.2.
It has a large number of mutations, especially amino-acid substitutions in spike. This pattern is characteristic of viruses that have evolved under antibody pressure in chronic human infections, with Omicron being the most prominent example.
For background on the original discovery of the variant, see the Pango GitHub issue, the lineage proposal GitHub issue, and early Twitter posts by Shay Fleishon and Ryan Hisner.
For real-time updated phylogenetic information on full BA.2.86 sequences, see this Nextstrain tree maintained by Cornelius Roemer and Richard Neher.
Also note there are reports of BA.2.86 in wastewater sequencing (see here and here).
In its spike protein, the new BA.2.86 variant has:
The number of spike amino mutations in the BA.2.86 variant relative to BA.2 and XBB.1.5 is comparable to number of mutations in first Omicron strains relative to Wuhan-Hu-1.
Notes: I670V is polymorphic among the sequences. The lack of R408S (relative to Wuhan-Hu-1) in Israeli sequence is being assumed to be an error. E554K is missing in the Israeli sequence, but this may be a sequencing artifact.
Estimates of effects of mutations from RBD deep mutational scanning by Tyler Starr, full-spike deep mutational scanning of BA.2 and XBB.1.5 by Bernadeta Dadonaite (Bloom lab), the Bloom lab RBD escape calculator informed by data from Yunlong Cao, and definition of the NTD supersite by Matthew McCallum & David Veesler. Experiments were performed in various genetic backgrounds and so there could be unmodeled epistasis. The * indicates mutations only in some sequences of the new variant.
These are only estimates of mutation effects from deep mutational scanning experiments.
K356T
Adds N-linked glycan to RBD at N354 by creating Asn-X-Ser/Thr motif.
Addition of glycan is a very effective way to block antibodies from binding an epitope.
Mutations that introduce a glycan at N354 have been used in multiple different immunogen design studies to mask this epitope on the RBD.
The K356T mutation sometimes arose in patients treated with sotrovimab.
delV483
This mutation deletes a residue in the RBD's receptor-binding motif.
Despite deletion being drastic change, Tyler Starr's yeast-display and our lab's (Bernadeta Dadonaite) full-spike deep mutational scanning show delV483 only moderately reduces ACE2 affinity (less than F486S/V that fixed in earlier variants)
Preliminarily, our full-spike deep mutational scanning suggests this mutation causes only a modest reduction in serum antibody neutralization in XBB.1.5.
Therefore, although this deletion has a functional & antigenic effect, that effect is moderate (no greater than that of some point mutations that have fixed in past).
Note receptor-binding motif deletions have occurred in antigenic evolution of CoV-229E.
P1143L
Our pseudovirus deep mutational scanning consistently finds that mutations at P1140 and P1143 in S2 improve viral entry by spike-pseudotyped lentiviral particles in multiple different variants (see Fig. 7 of Dadonaite et al (2023)).
However, mutations at these sites have been rare in actual human SARS-CoV-2 evolution. In above paper we speculated these mutations destabilize prefusion trimer at beginning of S2 stem helix, leading to more rapid cell entry in pseudoviruses but negatively impacting transmission.
It is therefore interesting that this new variant, which has undergone at least limited human transmission, has this mutation.
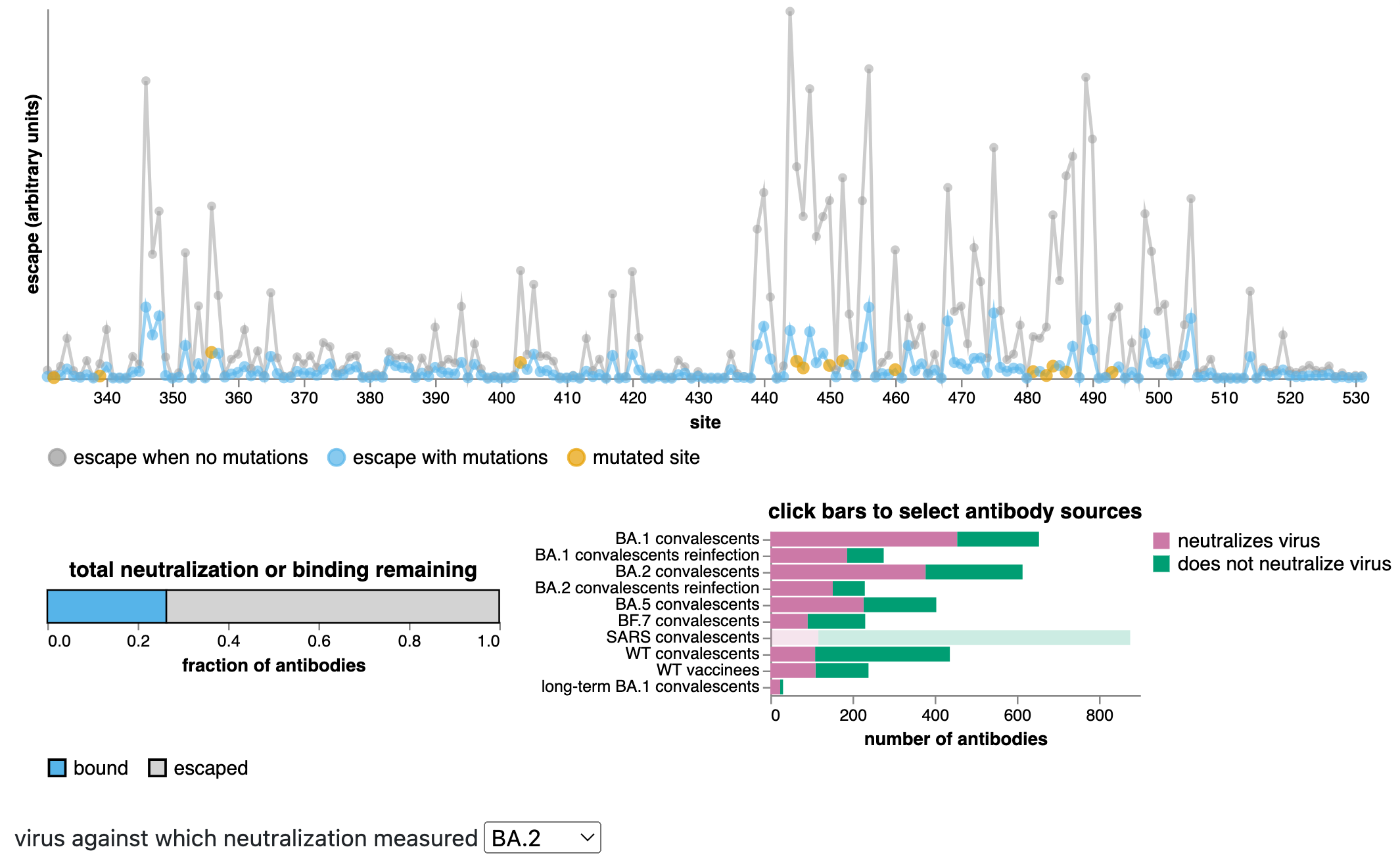
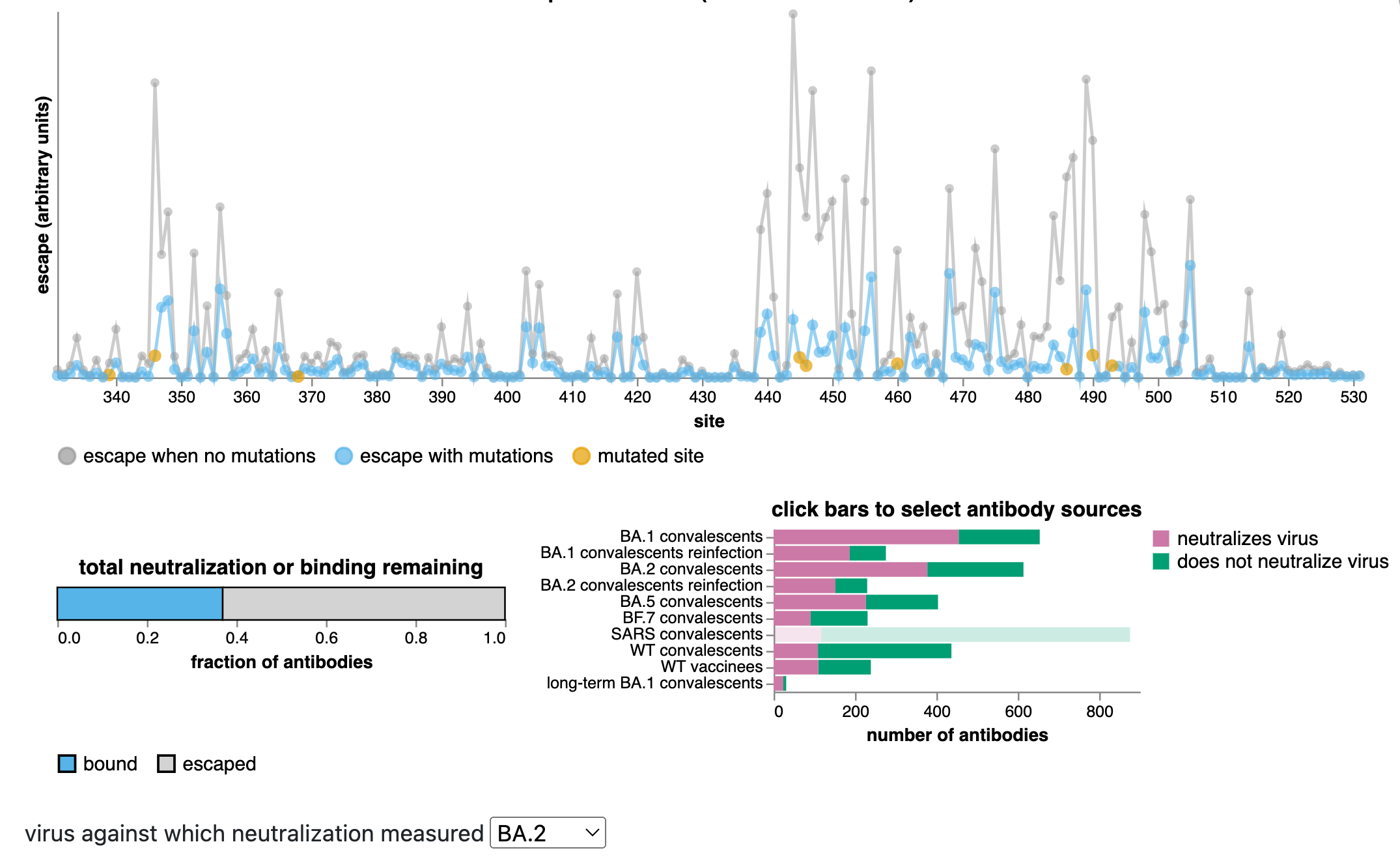
These estimates can be made using the antibody-escape calculator, which integrates deep mutational scanning data for thousands of human antibodies (data from Yunlong Cao's group).
The calculator allows us to estimate with fairly good accuracy how much mutating a set of sites will escape antibodies that neutralize a specific variant.
Predictions from RBD escape calculator with data from Yunlong Cao.
Antibody escape of new BA.2.86 variant relative to BA.2
Antibody escape of XBB.1.5 relative to BA.2
This comparison is informative as now many people have antibodies directed to XBB variants due to infection with XBB over the last half year.
Many mutations in new BA.2.86 variant relative to XBB.1.5 are same as those relative to BA.2, but new variant:
Estimates of effects of mutations from RBD deep mutational scanning by Tyler Starr, full-spike deep mutational scanning of BA.2 and XBB.1.5 by Bernadeta Dadonaite (Bloom lab), the Bloom lab RBD escape calculator informed by data from Yunlong Cao, and definition of the NTD supersite by Matthew McCallum & David Veesler. Experiments were performed in various genetic backgrounds and so there could be unmodeled epistasis. The * indicates mutations only in some sequences of the new variant.
These are only estimates of mutation effects from deep mutational scanning experiments.
New BA.2.86 variant has many spike amino-acid mutations relative to its BA.2 parent, and is an evolutionary jump similar in size to that which originally gave rise to Omicron.
Many of these mutations are antigenically important, indicating BA.2.86 variant evolved under strong antibody selection.
Deep mutational scanning indicates BA.2.86 variant will have equal or greater escape than XBB.1.5 from antibodies elicited by pre-Omicron and first-generation Omicron variants. It also has antigenic mutations relative to XBB.1.5 (the strain for the fall vaccine), and so may have a further antigenic advantage relative to XBB variants now that a substantial fraction of the population has also been exposed to XBB variants.
To be successful, BA.2.86 variant would need to combine this antigenic advantage with inherent transmissibility close to current XBB variants. Currently there are insufficient data to accurately estimate BA.2.86's transmissibility, but the variant merits further high-priority monitoring for signs of spread.
While neutralizing antibodies (which are partially escaped by highly mutated variants like BA.2.86) provide best protection against infection, there are also broader mechanisms of immunity elicited by vaccination and infection that provide some protection against severe disease even for very heavily mutated variants. So even if a highly mutated new variant like BA.2.86 starts to spread, we will be in a better place than we were in 2020 and 2021, since most people have some immunity to SARS-CoV-2 now.
By Jesse Bloom
Analysis of spike mutations in new second-generation BA.2 variant with many mutations




