








Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Research Center / HHMI
Slides at https://slides.com/jbloom/sars-cov-2-omicron
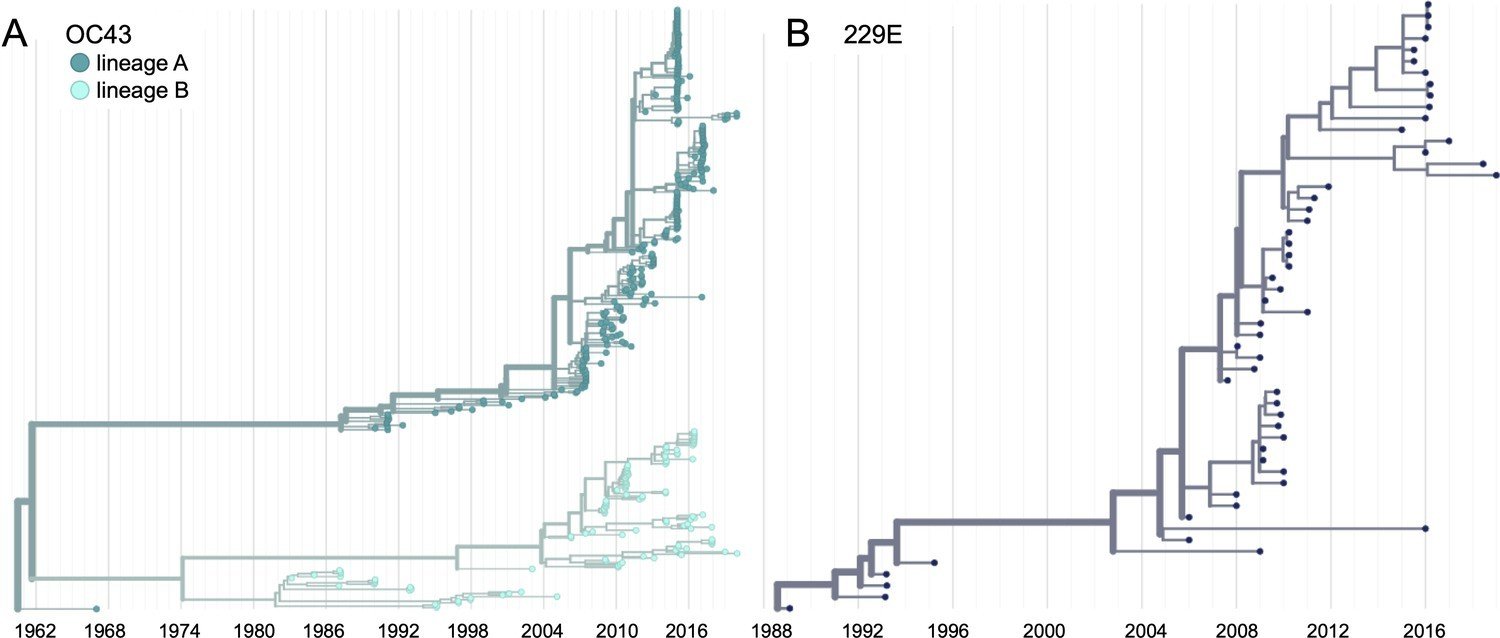
First let's look at another human coronavirus: CoV-229E causes common colds and has been circulating in humans since at least 1960s.
We experimentally generated CoV-229E spikes at ~8 year intervals so we could study them in the lab:
- 1984
- 1992
- 2001
- 2008
- 2016
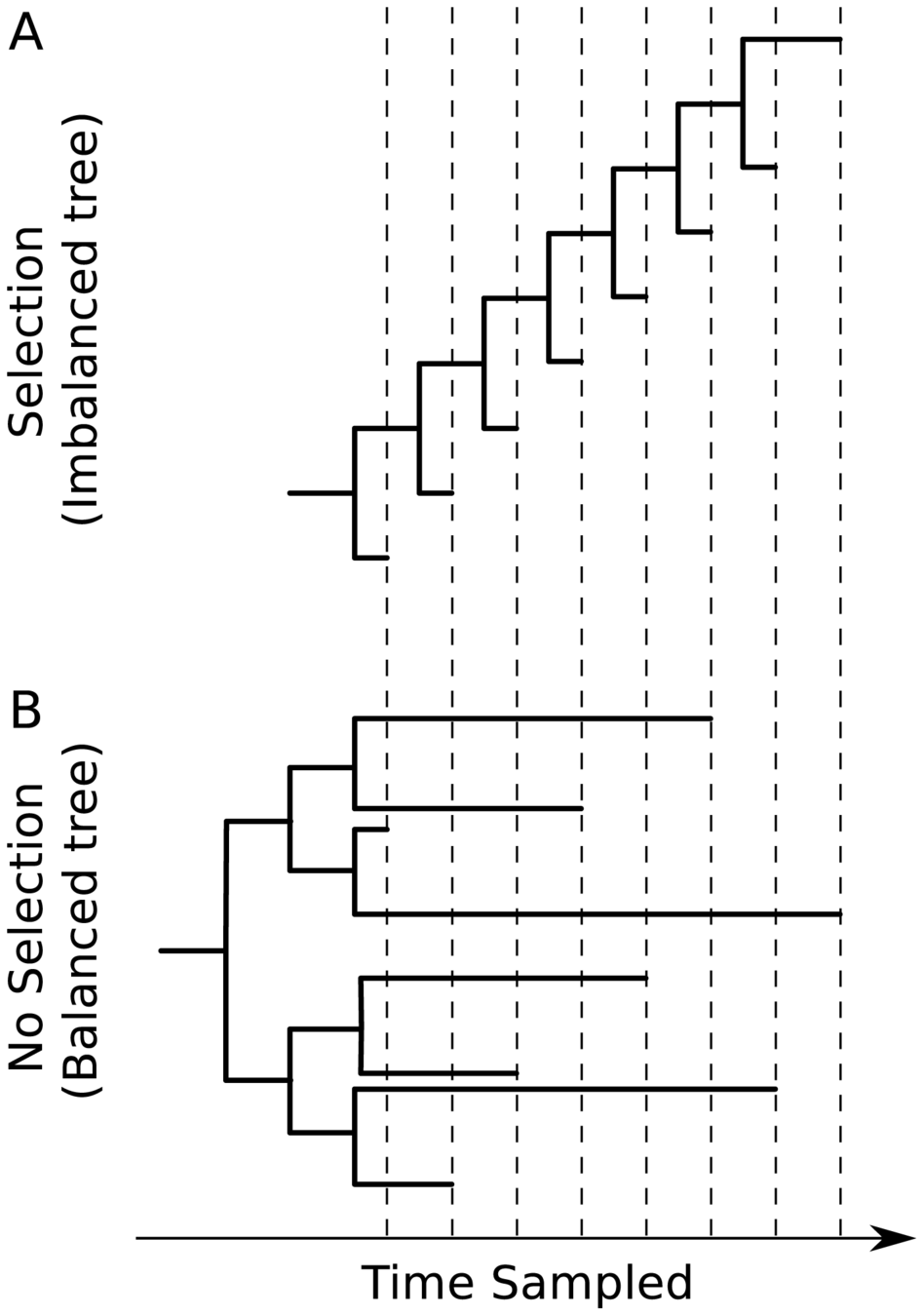
Note "ladder-like" shape of tree
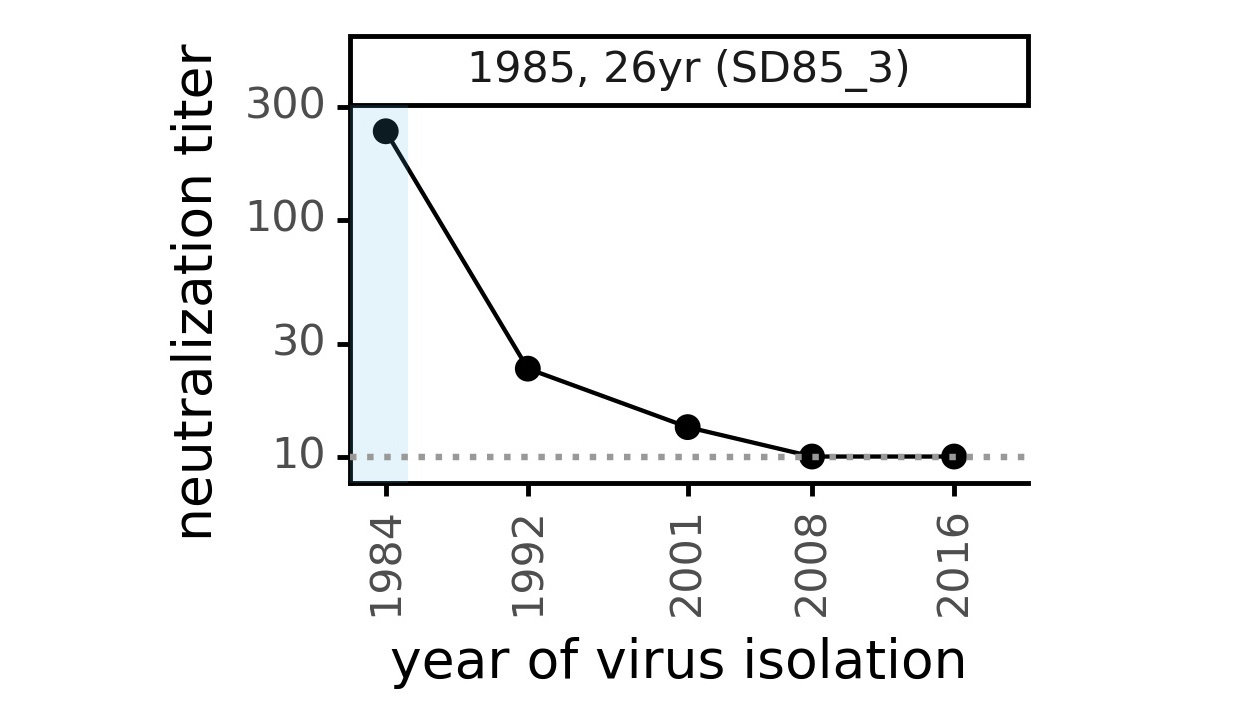
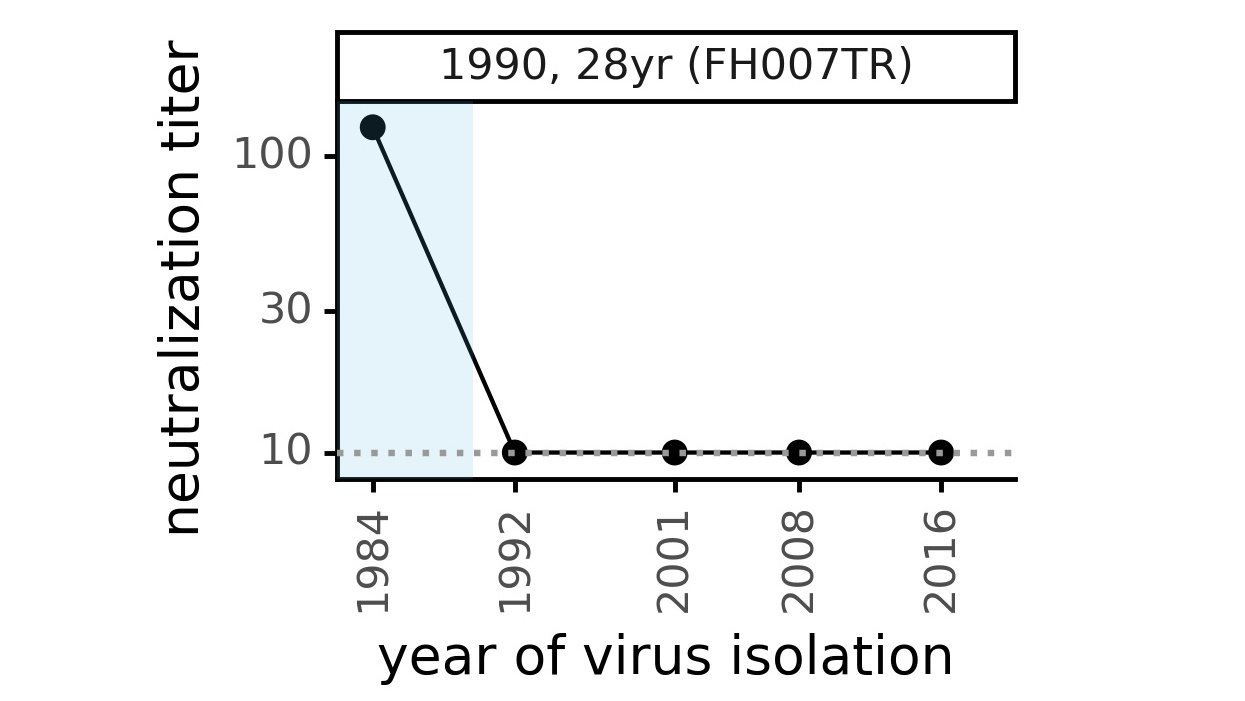
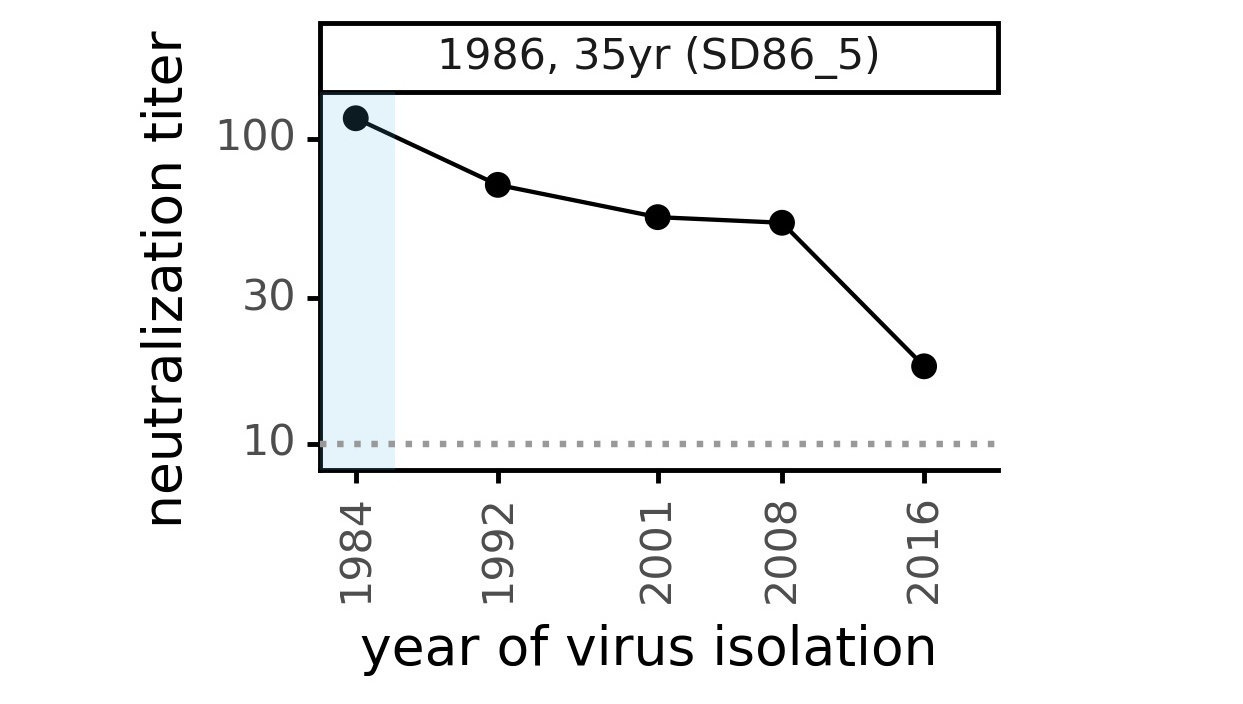
Serum collected in 1985 neutralizes virus with spike from 1984, but less effective against more recent viruses.
We are studying basis of these differences, as ideally vaccines would elicit more evolution-resistant sera as on the right.
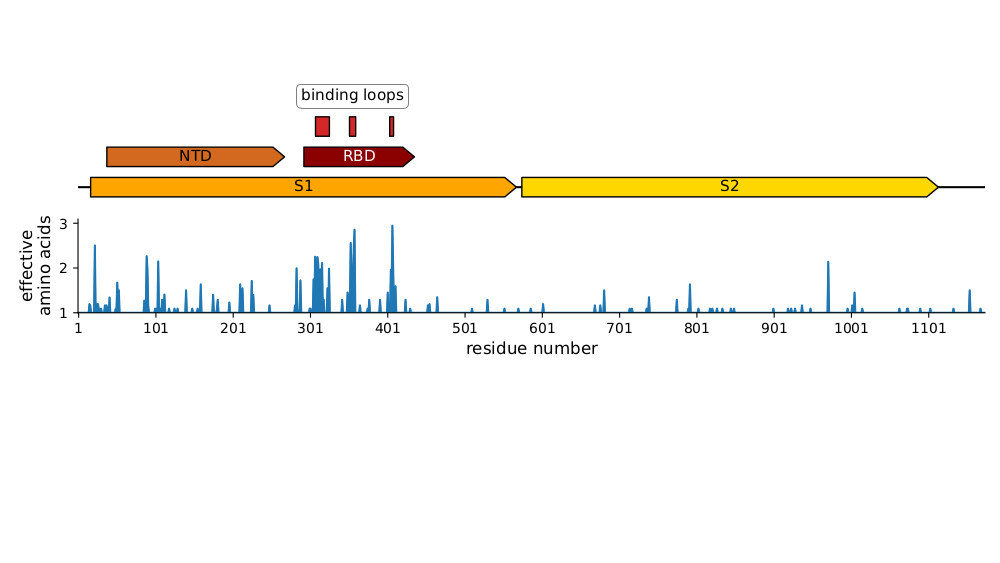
Plot of sequence variability across CoV-229E spike taken from Eguia, ..., Bloom, PLoS Pathogens (2021) . See also Wong, ..., Rini, Nature Communications (2017) and Li, ..., Rini, eLife (2019) for detailed structural studies of evolution in receptor-binding loops.
Similar to SARS-CoV-2 variants like Omicron except they also often have mutations that affect spike proteolytic processing.
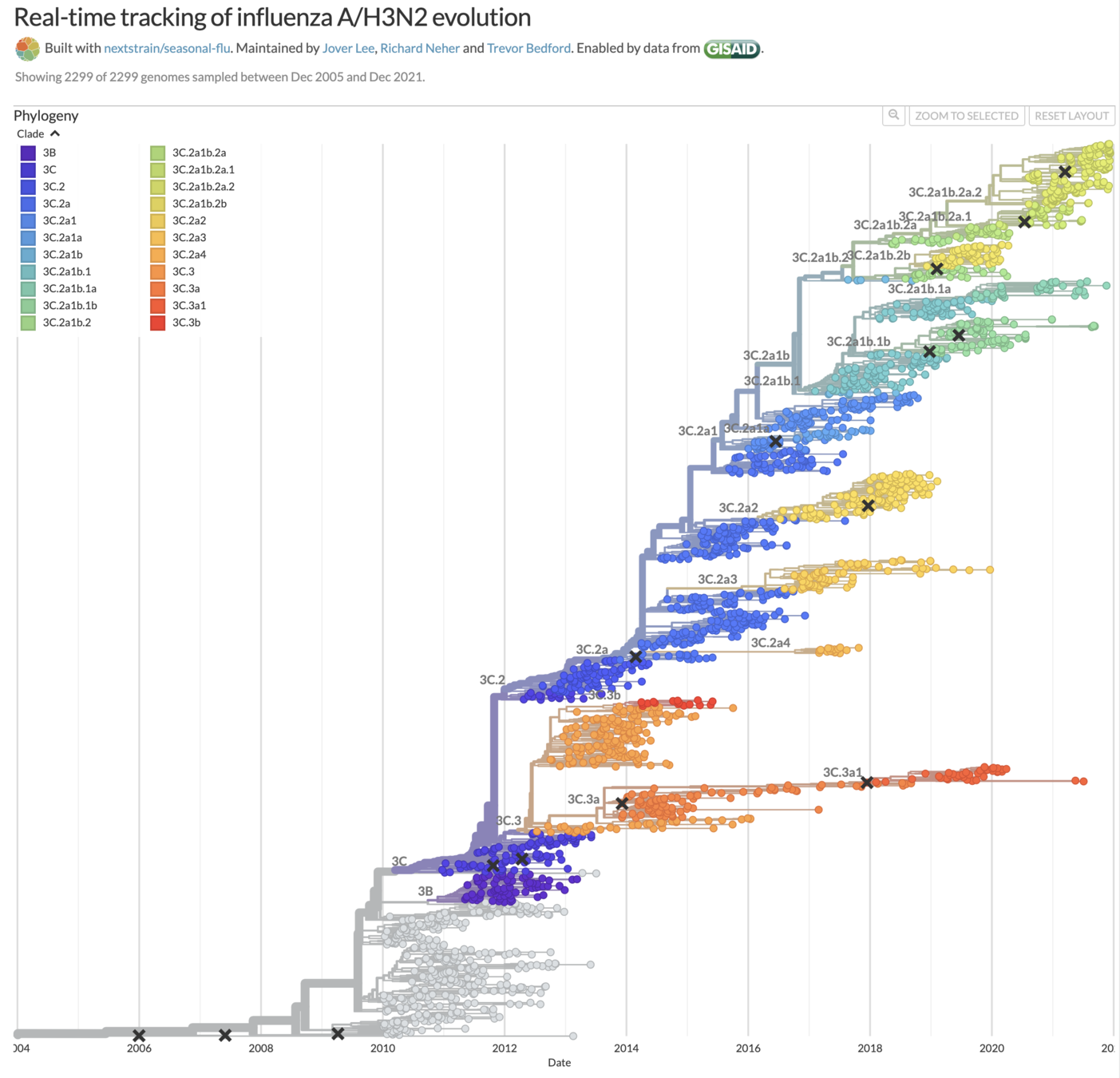
Taken from Volz et al (2013), Kistler and Bedford (2021), and Kucharski (Twitter, 2022)
Human respiratory viruses that undergo rapid antigenic evolution tend to have a "ladder-like" tree because they evolve under "winner-take-all" paradigm where most successful variant displaces others. Viruses that evolve without antigenic selection (or have high diversity over geographic space) have more balanced trees.
CoV-229E tree looks like this
Human H3N2 influenza also tends to have ladder-like trees, and in years where it hasn't that's made vaccine strain selection difficult (vaccine strains marked with x).
Taken from Volz et al (2013), Kistler and Bedford (2021), and Kucharski (Twitter, 2022)
Human CoV-OC43 has split into two lineages, although each lineage has a ladder-like tree. Something similar has happened with influenza B.
Taken from Volz et al (2013), Kistler and Bedford (2021), and Kucharski (Twitter, 2022)
Taken from Volz et al (2013), Kistler and Bedford (2021), and Kucharski (Twitter, 2022)
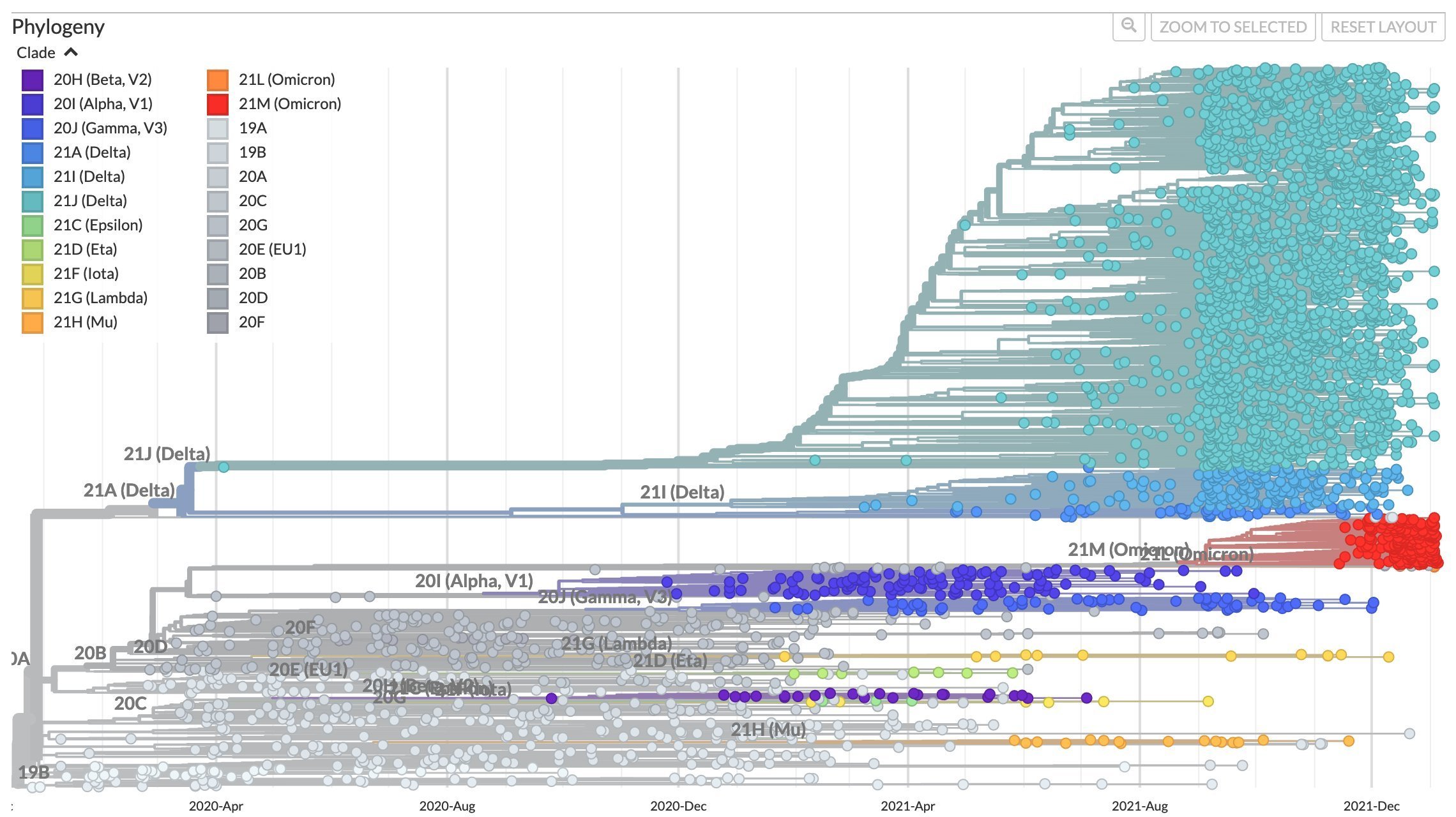
After early fixation of D614G, the SARS-CoV-2 tree has not been ladder-like
Other coronaviruses evolve antigenically with mutations in same regions as SARS-CoV-2 variants (exception is SARS-CoV-2 variants also have mutations that affect proteolytic processing, probably because initial virus was poorly adapted to its furin-cleavage site).
Other antigenically evolving human respiratory viruses have ladder-like phylogenetic trees, which is key to enabling vaccine updates as it provides some level of predictability (the next variant usually is descended from the last one).
So far SARS-CoV-2 has mostly defied ladder-like paradigm: Omicron not descended from Delta; Delta not descended from Alpha. However, it's still early and right now we are seeing combined effect of selection for antibody escape and increased transmissibility--presumably transmissibility will eventually plateau. Despite having been wrong in thinking next variant would come from Delta, I still think evolution will eventually become ladder-like.
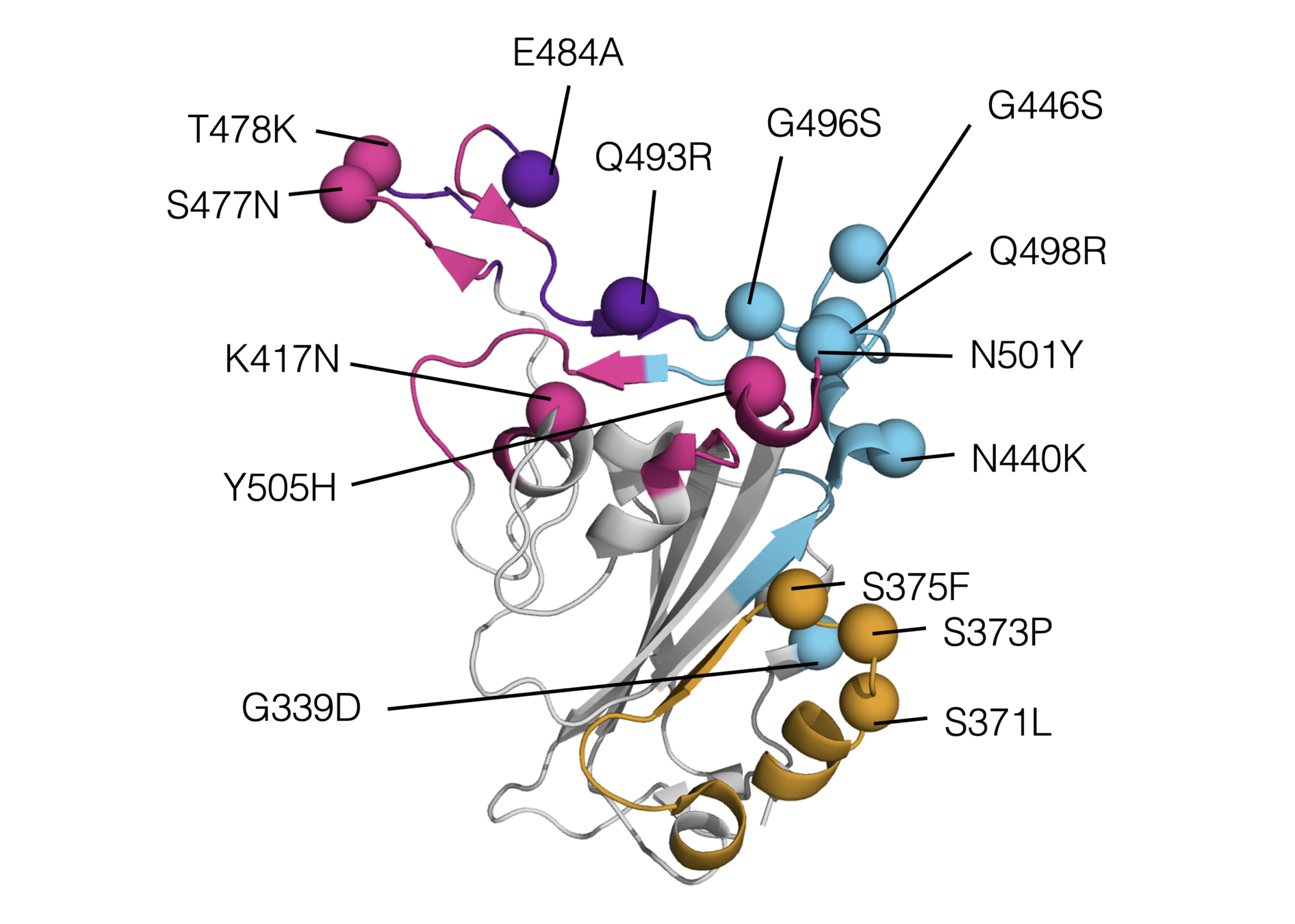
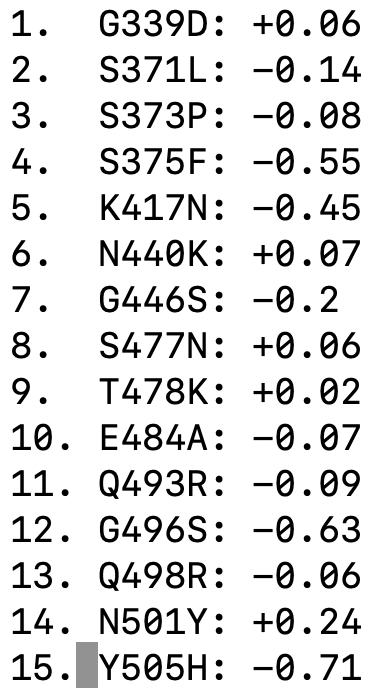
We have used deep mutational scanning to measure how all individual mutations in "original" (Wuhan-Hu-1) RBD affect ACE2 affinity: two-thirds of mutations in Omicron's RBD are individually deleterious for ACE2 affinity
Note that above refers to RBD binding affinity. Full spike binding also influenced by up-down RBD conformational balance, which may differ in Omicron
Two studies have shown that Q498R and N501Y combine to lead to a much greater increase in affinity than either alone. See this Twitter thread for more evolutionary details.
It is also possible that there is epistasis among other combinations of mutations in Omicron, although this has not yet been experimentally elucidated.
Omicron represents first major example where the effects of combined RBD mutations could not be predicted fairly well from single mutants.
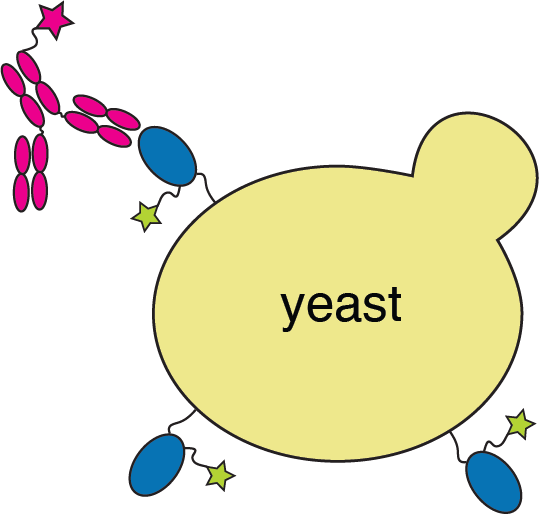
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
See Seth Zost's thread for a good summary of therapeutic antibodies and Omicron
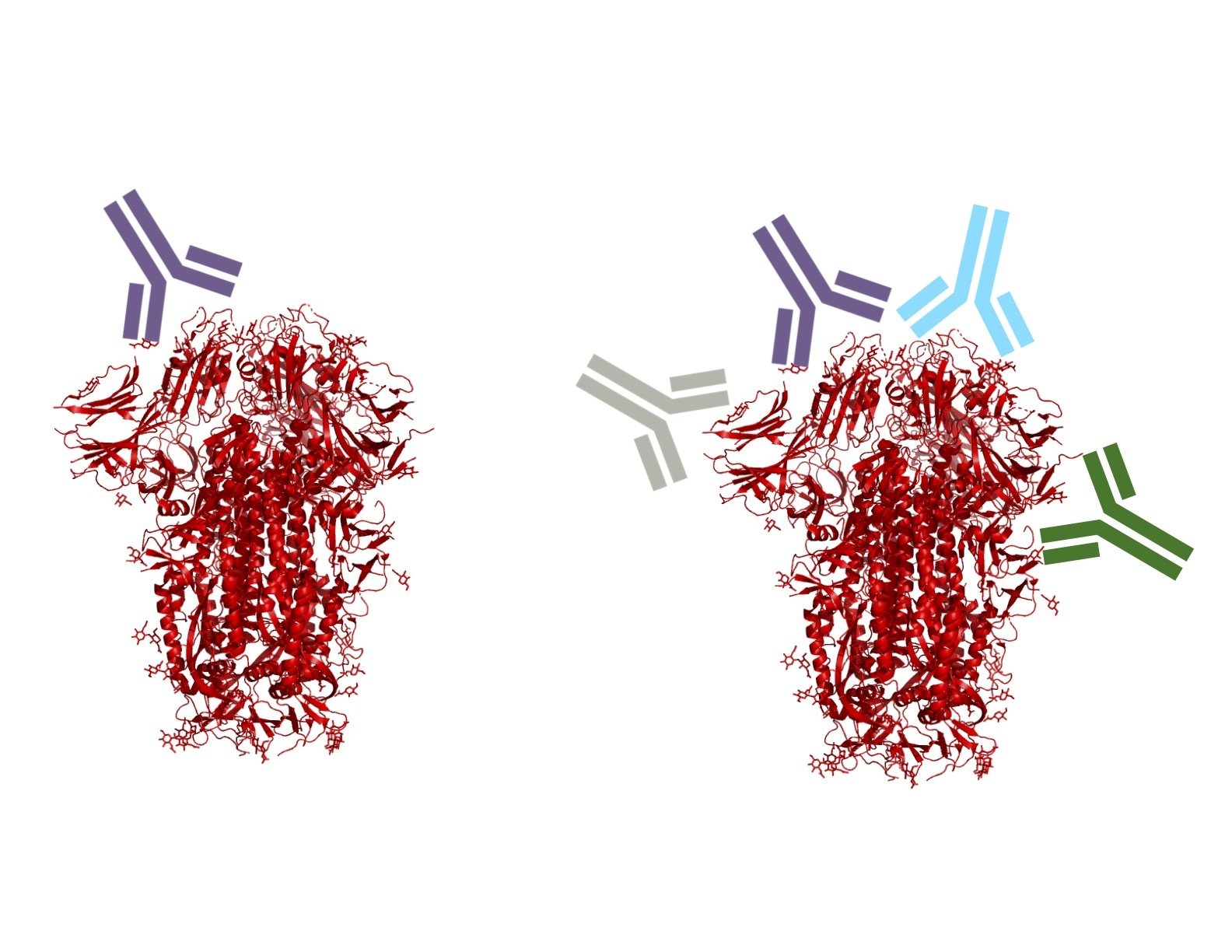
Monoclonal antibodies bind one epitope, so can usually be escaped by single mutation
Polyclonal antibodies can bind many epitopes, so often more resistant to escape
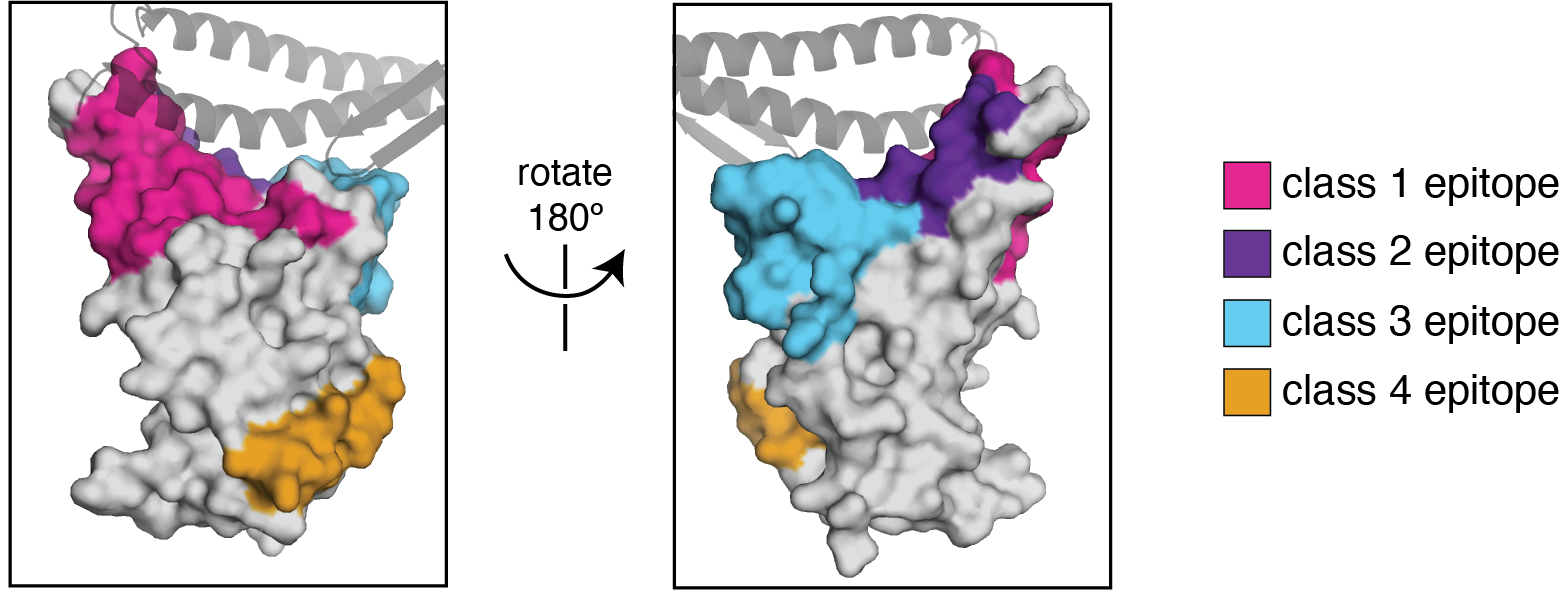
We use the antibody classification scheme from Barnes et al Nature (2020). The extension of these antibody classes to escape mapping is in Greaney et al (2021), and escape maps are available here. Class 1, 2, and 3 antibodies are often potently neutralizing, while class 4 antibodies are less neutralizing (see: Piccoli et al (2020), Dejnirattisai et al (2021), Liu et al (2020), Zost, et al (2020)).
Toy example of calculating escape from polyclonal mix:
https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/mini-example-escape-calc/
Escape-calculator with deep mutational scanning data from many antibodies:
https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
Predictions correlate well with neutralization assays:
https://jbloomlab.github.io/RBD_escape_calculator_paper/neut_studies.html
See Greaney et al (2021) for paper on escape-calculator, and here for more examples on usage.
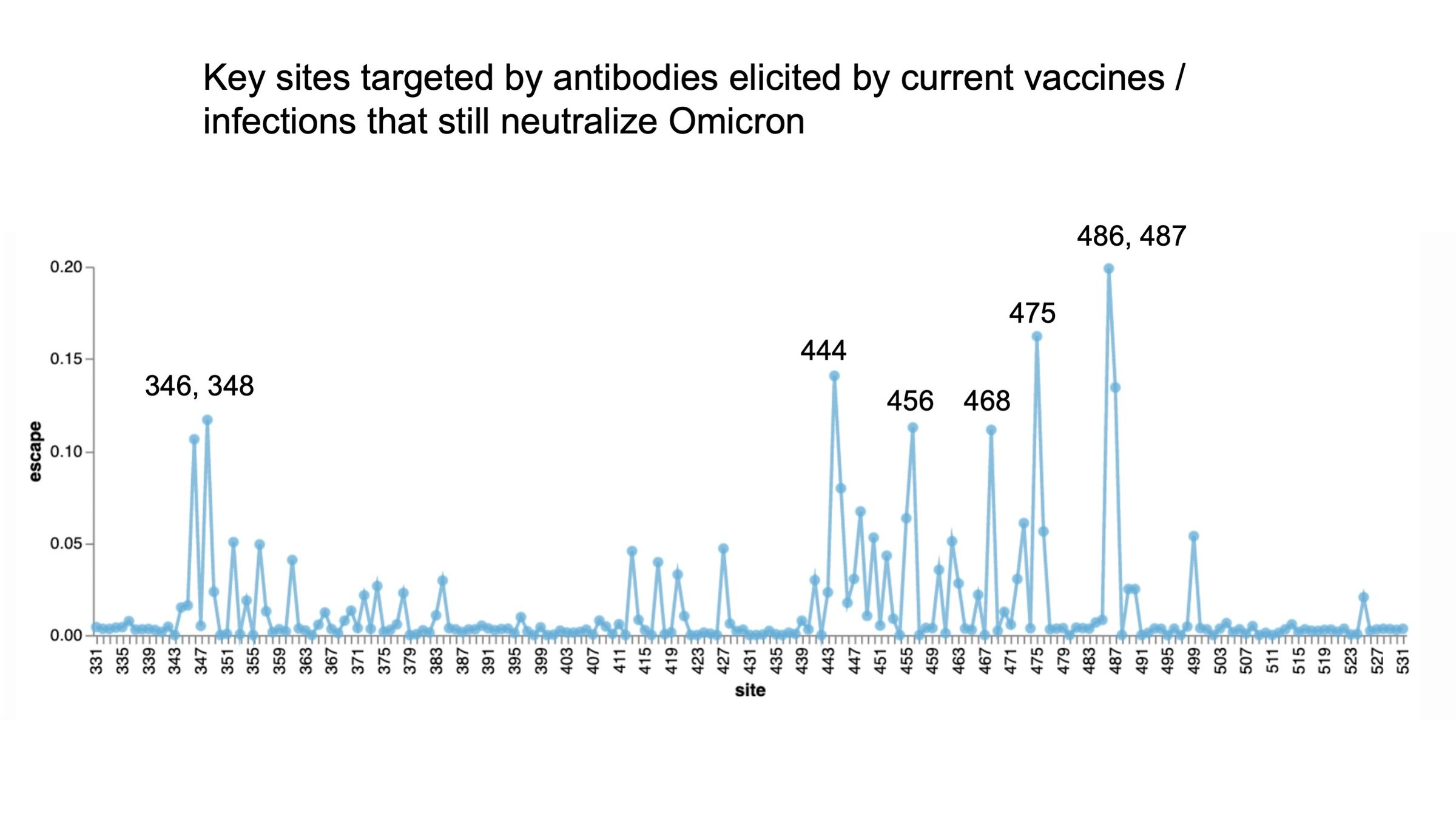
Omicron has a lot more escape than other variants.
Sites to watch for future antigenic mutations include 346.
Crowe lab (Vanderbilt)
Chu lab (Univ Wash)
Veesler lab (Univ Wash)
King lab (Univ Wash)
Li lab (Brigham & Women's)
Boeckh lab (Fred Hutch)
Alex Greninger (Univ Wash)
Nussenzweig lab (Rockefeller)
Bjorkman lab (Caltech)
Tyler Starr
Allie Greaney
Rachel Eguia
Bloom lab (Fred Hutch)
Sarah Hilton
Kate Crawford
Andrea Loes
By Jesse Bloom
Interpreting the evolution of SARS-CoV-2 in the context of Omicron



