






















Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/sars2-uga
I am on the scientific advisory boards of Apriori Bio and Oncorus
I have consulted on topics related to viral evolution for Moderna and Merck
I am an inventor on Fred Hutch licensed patents related to deep mutational scanning of viral proteins
My lab has unfunded research collaborations with Vir Biotechnology
From 1918 to 1957, H1N1 influenza circulated in humans.
In 1957, H1N1 disappeared from humans due to new H2N2 pandemic.
In 1977, old H1N1 strain from ~1954 was accidentally re-released (probably during misguided vaccine trial) and caused a global pandemic.
"One boy from Hong Kong had a transient febrile illness from 15 to 18 January. On Sunday 22 January, three boys were in the college infirmary… 512 boys (67%) spent between three and seven days away from class."
"Of about 130 adults who had some contact with the boys, only one, a house matron, developed similar symptoms."
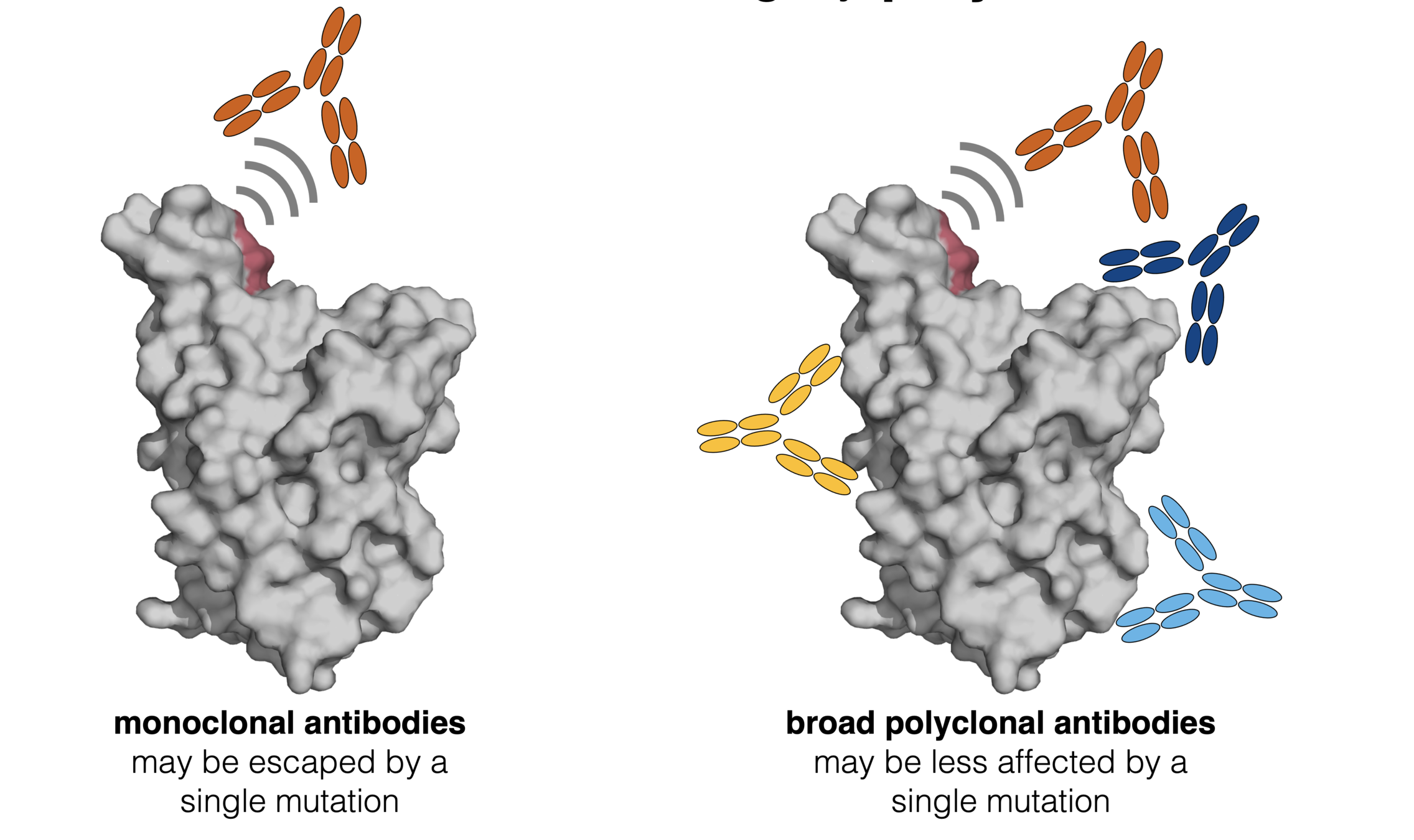
Why some viruses evolve to escape immunity while others don't is a deep question outside scope of this talk. See here for some possible explanations.
Rate of viral antigenic evolution
Measles
Influenza
Rate of viral antigenic evolution
Measles
Coronaviruses
???
???
Influenza
Rate of viral antigenic evolution
Measles
Coronaviruses
???
???
Influenza
We decided to study another human coronavirus: CoV-229E causes common colds and has been circulating in humans for a long time.
We experimentally generated CoV-229E spikes at ~8 year intervals so we could study them in the lab:
- 1984
- 1992
- 2001
- 2008
- 2016
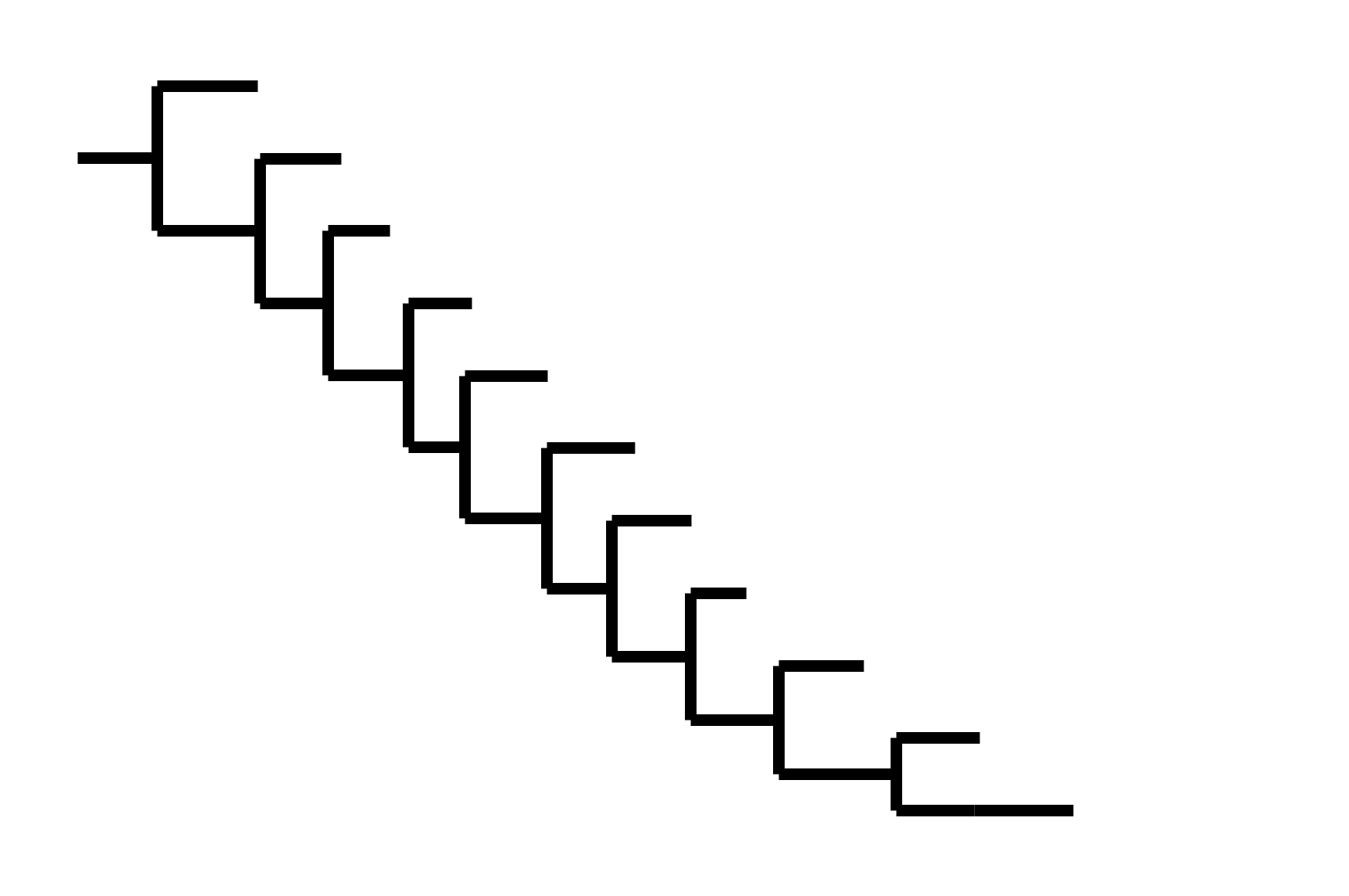
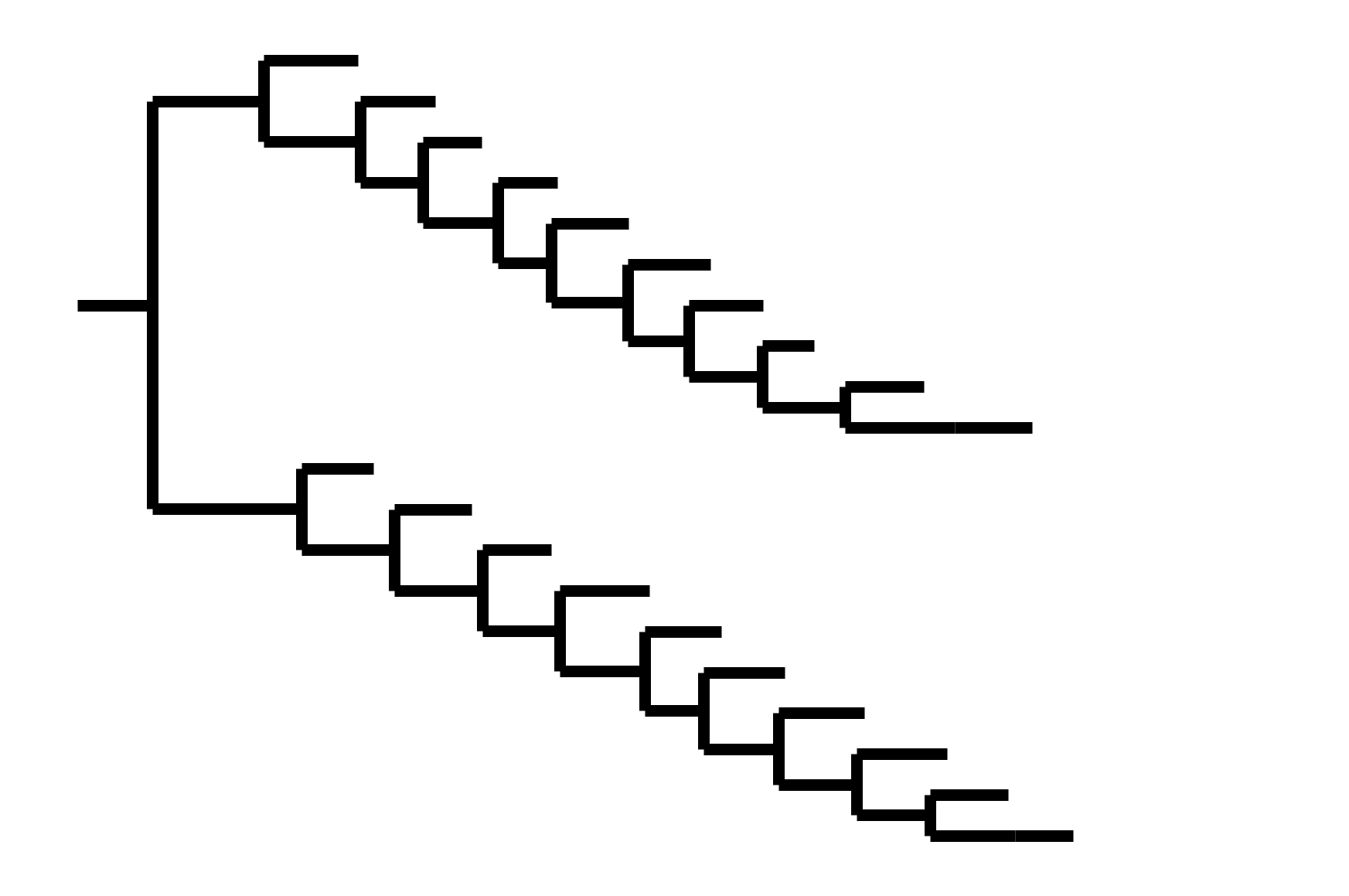
Note "ladder-like" shape of tree
Serum collected in 1985 neutralizes virus with spike from 1984, but less effective against more recent viruses.
We are studying basis of these differences, as ideally vaccines would elicit more evolution-resistant sera as on the right.
Rate of viral antigenic evolution
Measles
CoV-229E
CoV-OC43
SARS-CoV-2
Influenza
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
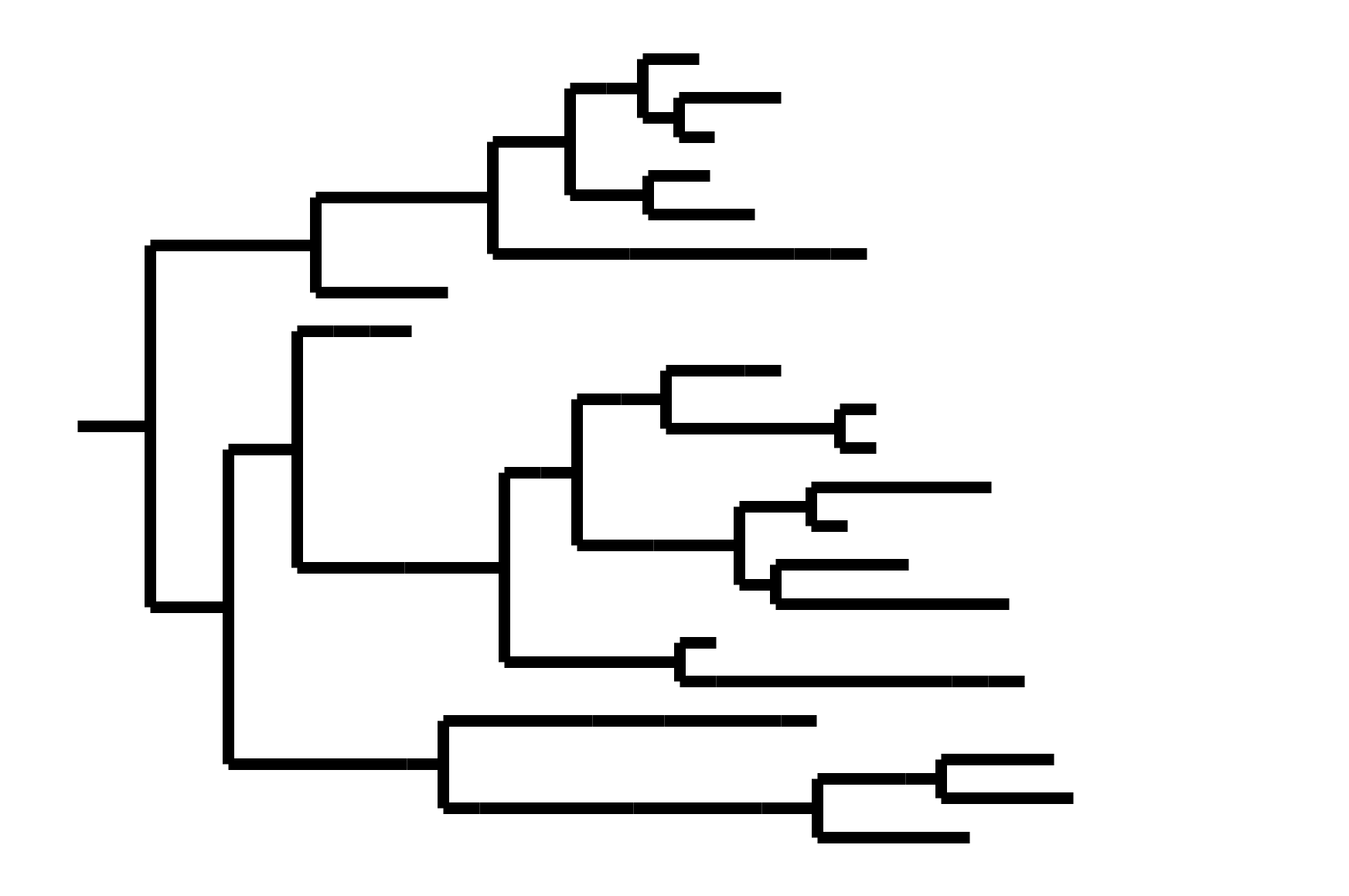
CoV-OC43 split into two ladder-like lineages. Influenza B evolves this way too. It's theoretically possible to pick well-matched bivalent vaccine.
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
CoV-OC43 split into two ladder-like lineages. Influenza B evolves this way too. It's theoretically possible to pick well-matched bivalent vaccine.
In non-ladder-like tree, next variant not descended from recent successful one. Makes picking vaccine strains difficult.
For each Nextstrain clade, number of mutations plotted versus date of clade origin, with trend line fit only to non-Omicron clades. Omicron clades are circled in red.
Data extracted from Neher (2022) and re-plotted.
See here for more details on why Omicron probably evolved in a chronic human infection.
A substantial amount of the evolution so far has been driven by increases in inherent transmissibility.
However, we are increasingly seen evolution driven by immune escape.
Human endemic viruses probably eventually plateau inherent transmissibility and evolve mostly to escape immunity. So the increased-transmissibility aspects of SARS-CoV-2 evolution may be transient features of the first few years.
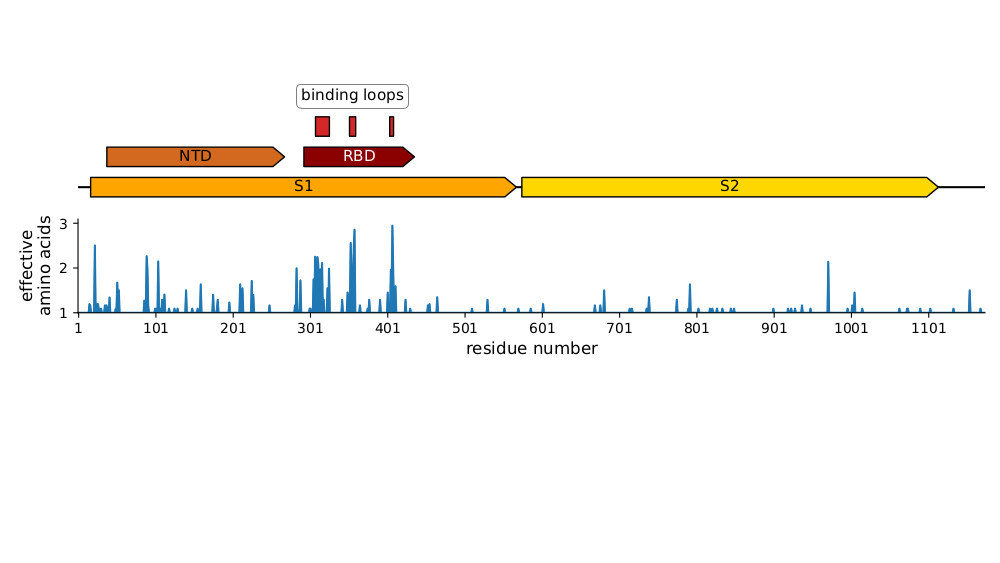
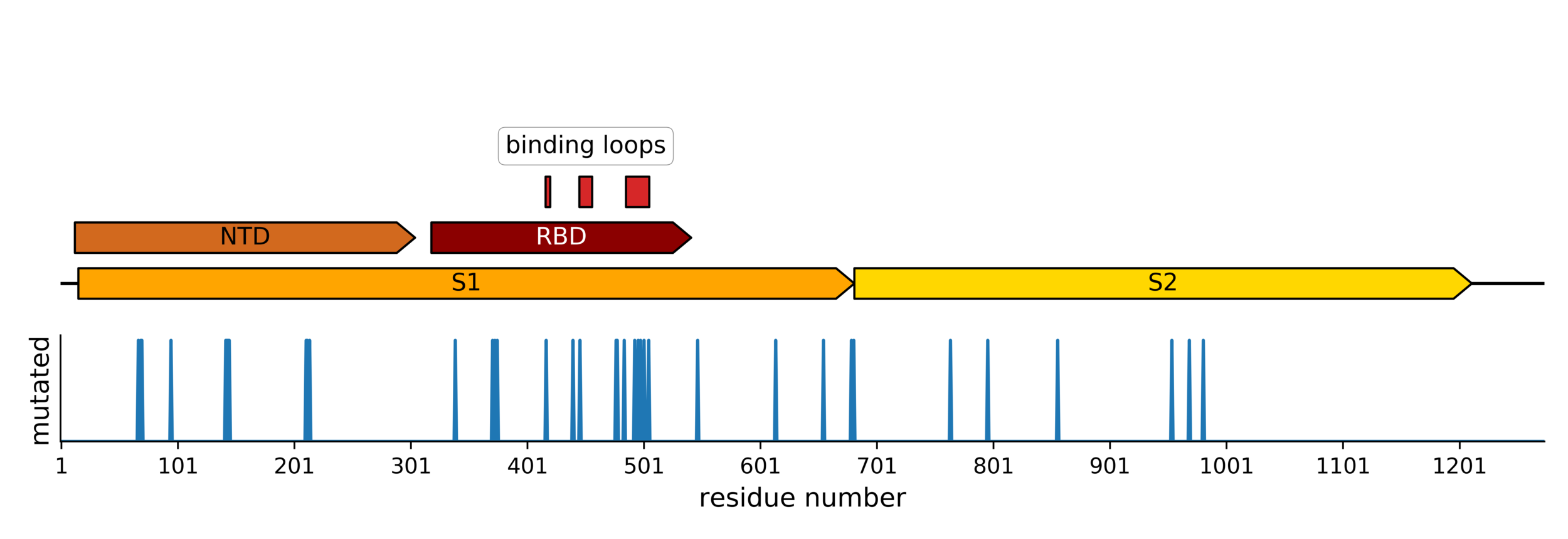
Sites of evolutionary change in the spike of CoV-229E over the last four decades
Sites of evolutionary change in the spike of CoV-229E over the last four decades
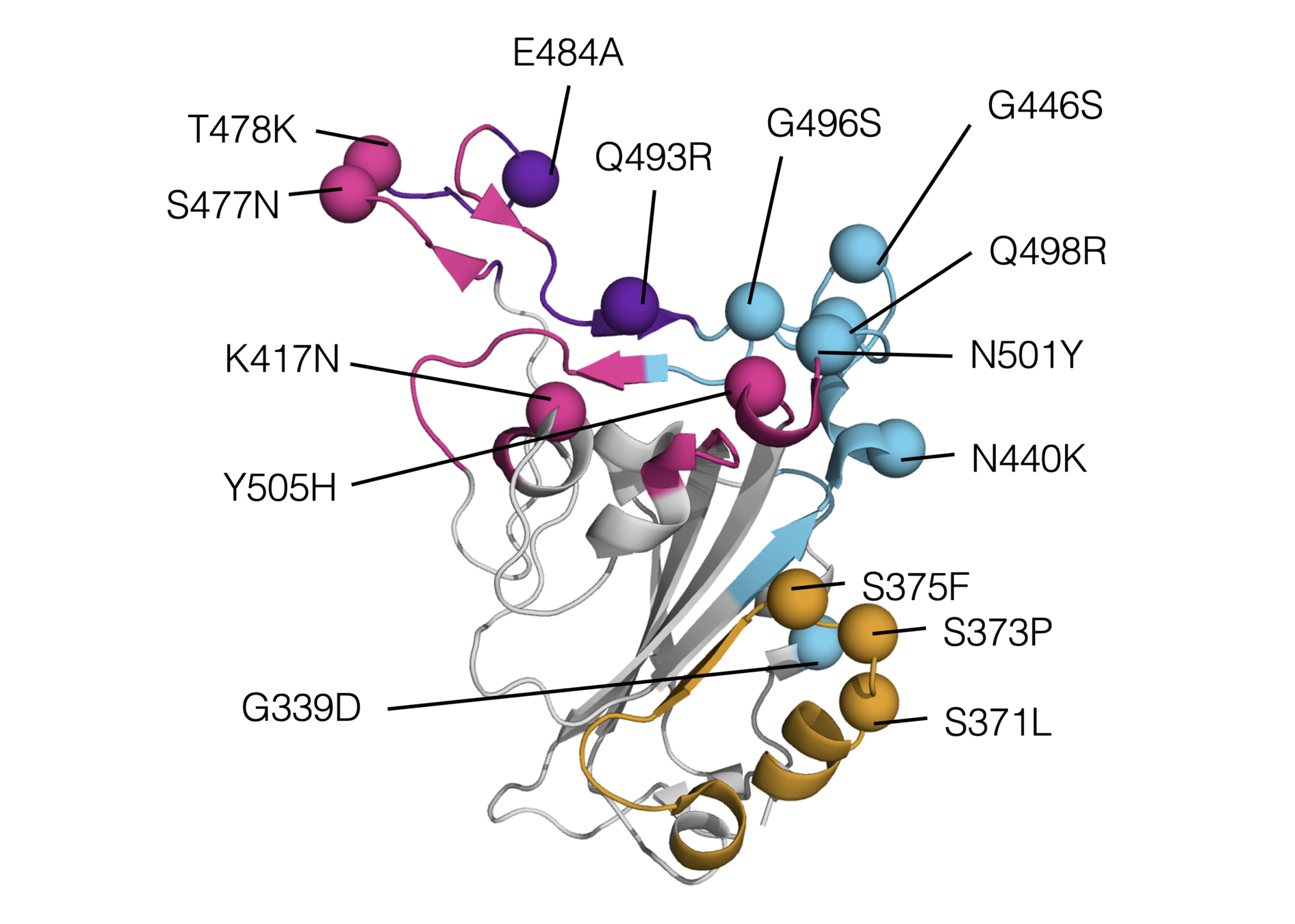
Sites of mutations in SARS-CoV-2 Omicron (BA.1) spike relative to Wuhan-Hu-1
Main difference is SARS-CoV-2 also fixing transmissibility-enhancing spike mutations that affect proteolytic processing and stabilize defects cause by furin-cleavage site
Human CoVs, which evolve to escape transmission-blocking immunity, show strongest selection in RBD.
So virus is telling us RBD antibodies matter most for blocking transmission. But other antibodies and T-cells still matter, and may reduce disease severity while putting less selection on virus.
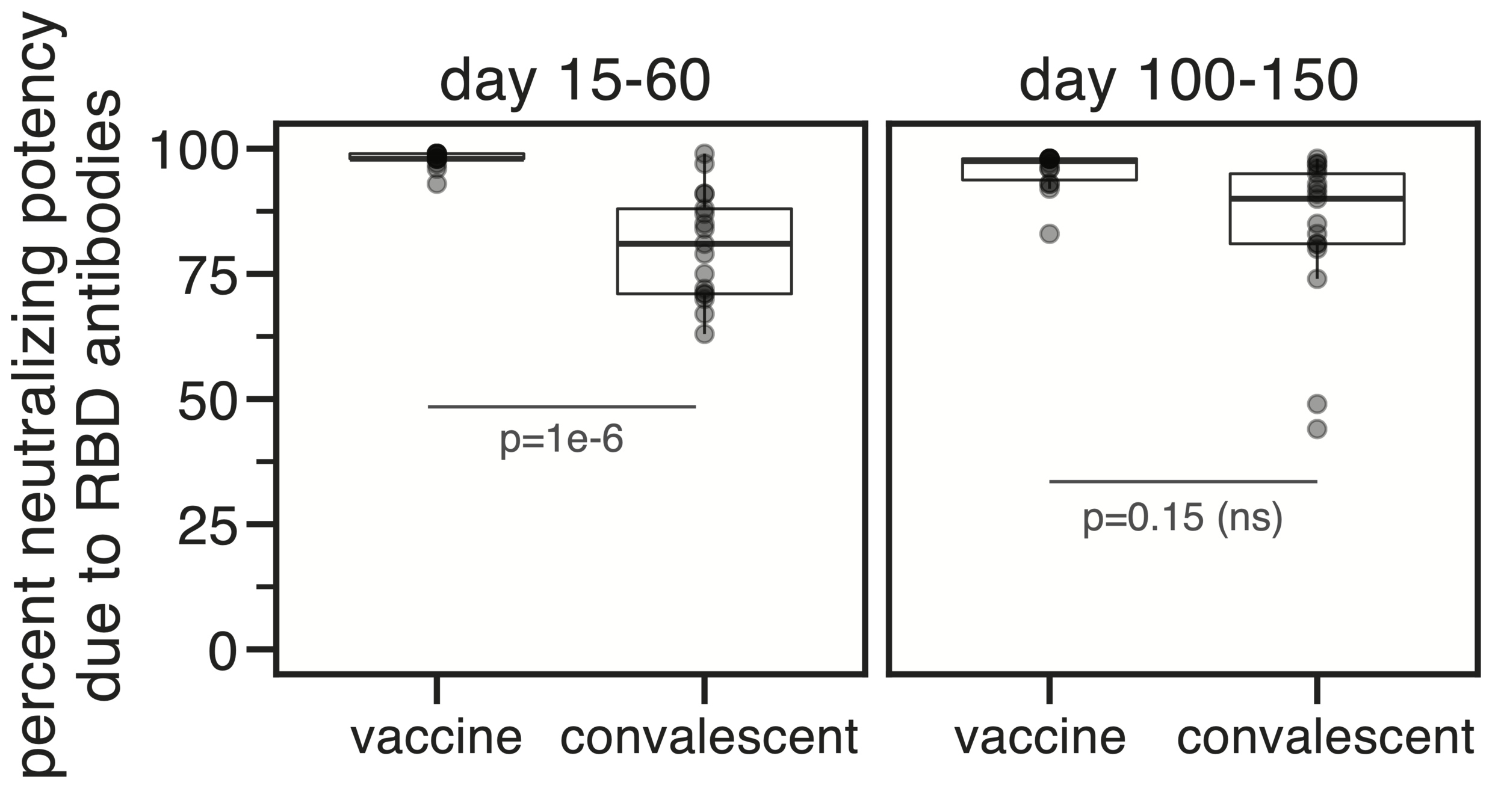
Most neutralizing activity from RBD antibodies (although antibodies to other domains can also be neutralizing).
RBD mutations in Omicron BA.1
RBD
fluorescent ACE2
yeast
fluorescent tag on RBD
Importantly, we use ACE2 titrations to measure true affinities, not just relative FACS binding signal; see here for details.
Library of yeast each expressing a different RBD mutant. Click here for details on how library is made.
Interactive heatmaps are available here, and are from Starr et al, 2022 and unpublished work.
Mutations in Omicron have net negative effect on ACE2 binding if summed as single mutants
Mutations in Omicron have net negative effect on ACE2 binding if summed as single mutants
However, two mutations (Q498R & N501Y) work together so net effect ~zero when both present
25 of 31 residues in CoV-229E RBD that contact receptor varied during virus's evolution in humans over last ~50 years (Li et al, 2019)
There are lots of mutations to SARS-CoV-2 RBD that retain (and sometimes even enhance) ACE2 affinity (Starr et al, 2020; Starr et al, 2022)
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
Experiments combine flow cytometry and deep sequencing of a library of yeast expressing all RBD mutants
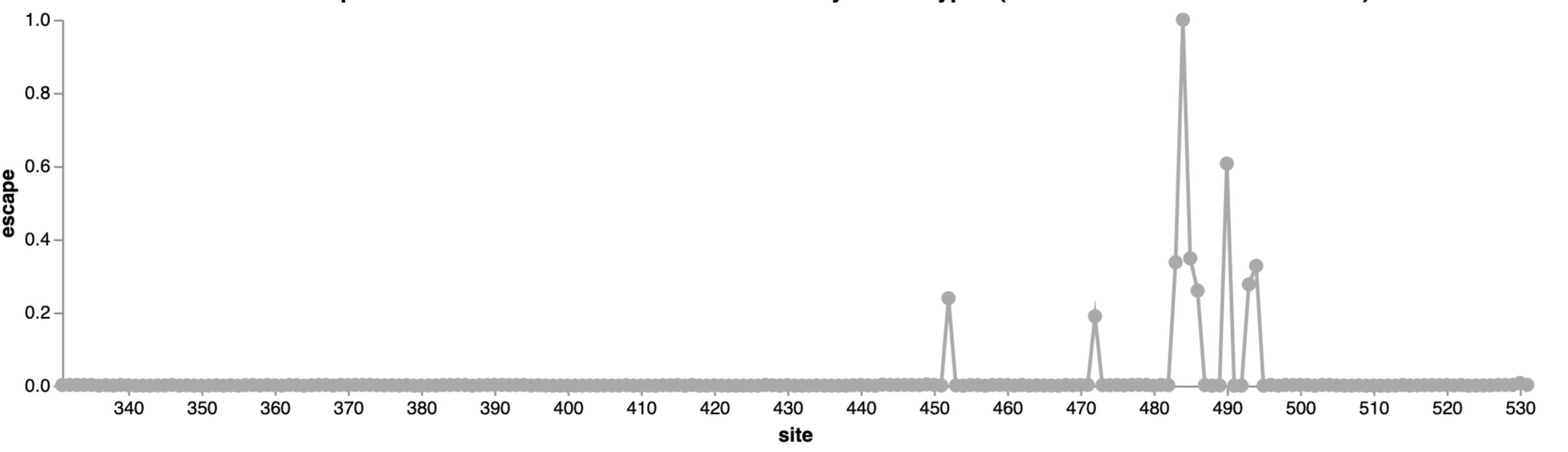
For interactive escape map, see: https://jbloomlab.github.io/SARS-CoV-2-RBD_MAP_LY-CoV555/
484
452
490
Interactive version of this mini example is at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/mini-example-escape-calc/
LY-CoV555 is escaped at both sites 484 and 490, so mutating either site has same overall effect
Average escape across all antibodies
Interactive version of this mini example is at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/mini-example-escape-calc/
Escape calculator is described in Greaney et al (2022), and is available at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
36 antibodies mapped by Tyler Starr & Allie Greaney in Bloom lab, from early SARS-CoV-2 strains
Escape calculator is described in Greaney et al (2022), and is available at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
36 antibodies mapped by Tyler Starr & Allie Greaney in Bloom lab, from early SARS-CoV-2 strains
1,522 (!) antibodies mapped by Sunney Xie, Richard Cao, Fanchong Jian, et al at Peking University. From early strains, BA.1, & patients with prior SARS-CoV-1 infection. See here.
417
446
484
417
446
484
417
446
484
486 is largest site of escape for antibodies not already escaped by mutations in BA.2
346
444-446
452
450
486
499
mutated in BA.4/BA.5
mutated in BA.2.75
346-348
444-446
356
Differences in antibody-escape mutations between people +/- BA.1 breakthrough infections is consistent with prior studies on other viruses showing that people with different exposure histories have different viral escape mutations.
452
486
mutated in BA.4/BA.5
mutated in BA.2.75
Crowe lab (Vanderbilt)
Chu lab (Univ Wash)
Veesler lab (Univ Wash)
King lab (Univ Wash)
Li lab (Brigham & Women's)
Boeckh lab (Fred Hutch)
Alex Greninger (Univ Wash)
Nussenzweig lab (Rockefeller)
Bjorkman lab (Caltech)
Katie Kistler (Fred Hutch)
Tyler Starr
Allie Greaney
Rachel Eguia
Bloom lab (Fred Hutch)
Sarah Hilton
Kate Crawford
Andrea Loes
These slides at:
By Jesse Bloom
Interpreting the evolution of SARS-CoV-2




