
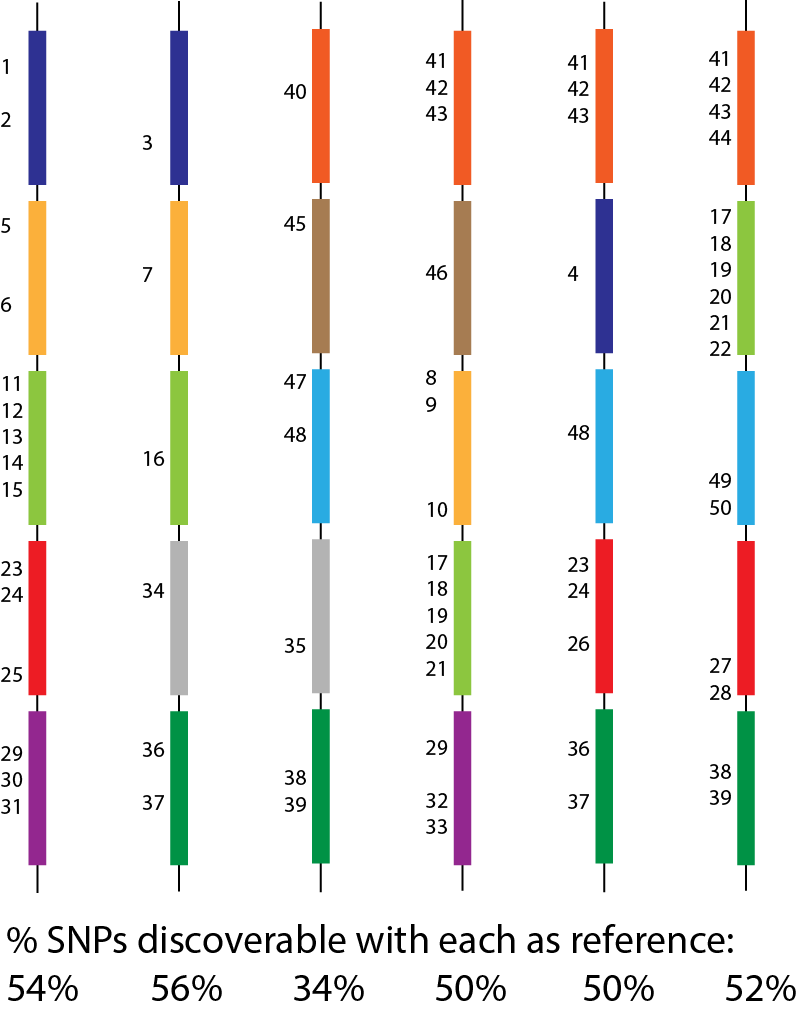










Michael Hall
I am a Bioinformatics PhD student in Zam Iqbal’s lab at EMBL-EBI. I currently work on using nanopore data and genome graphs to better call variation in bacterial genomes and to compare pan-genomes.
Michael B. Hall
Email: michael.hall@ebi.ac.uk
Second Year Report
Thesis Advisory Committee:
Zamin Iqbal - EMBL-EBI
John Marioni (Chair) - EMBL-EBI
Georg Zeller - EMBL Heidelberg
Estée Török - University of Cambridge
Genomics is now ubiquitous in clinical and public health microbiology. However, many significant challenges remain:
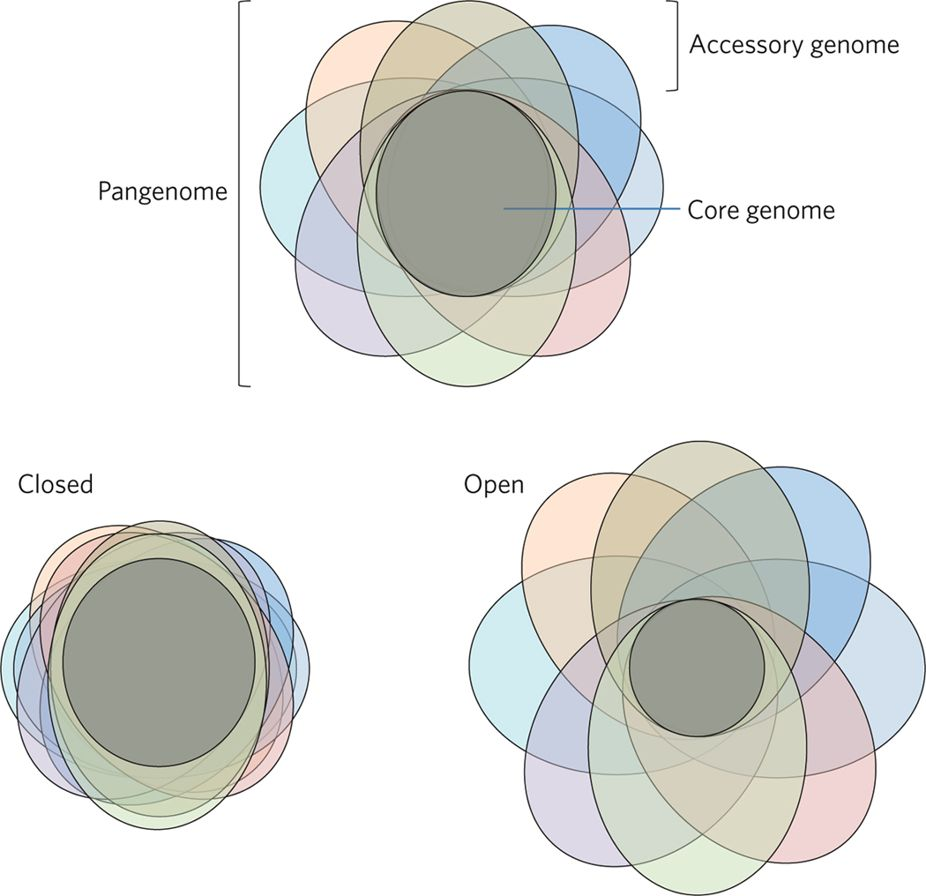
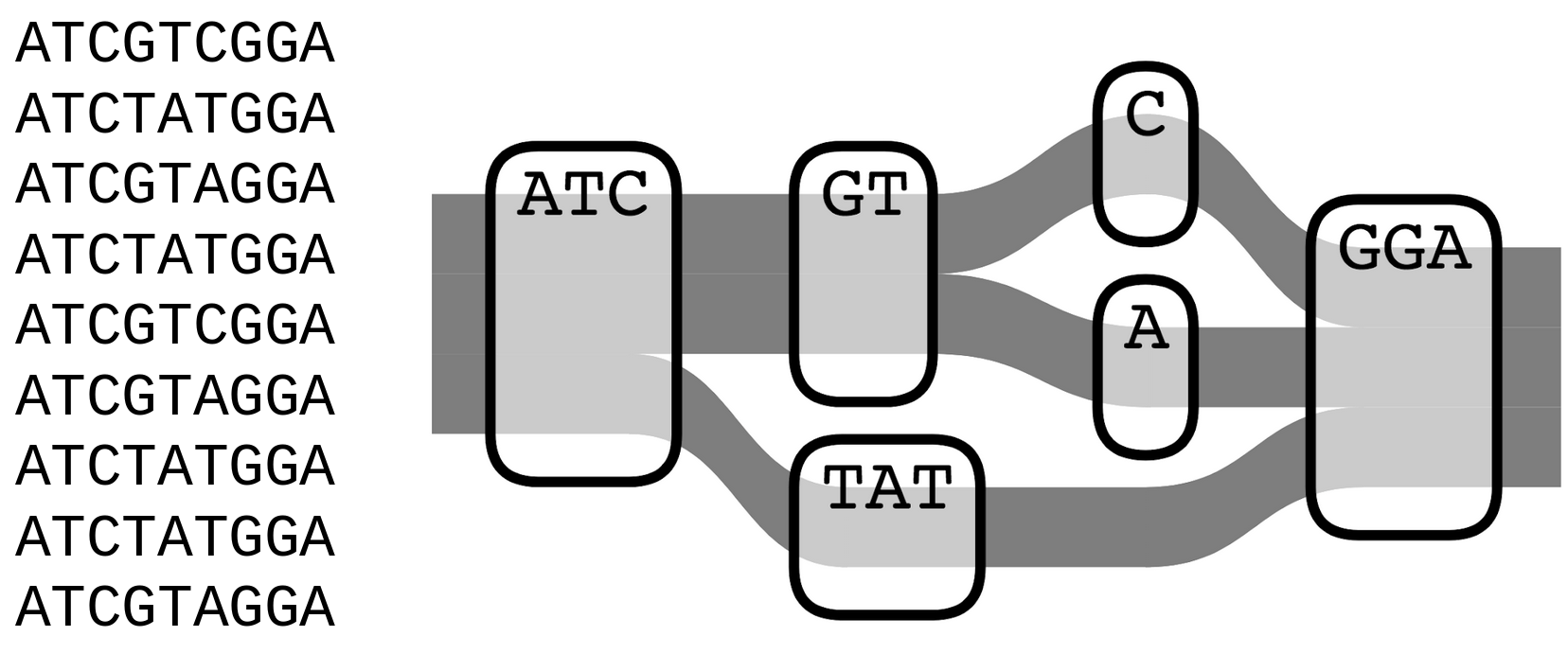
• Bacterial genomes harbour vast amounts of diversity, even within a species, and traditional reference-based approaches are problematic.
• Much of the variation in bacteria is fundamentally inaccessible to short reads.
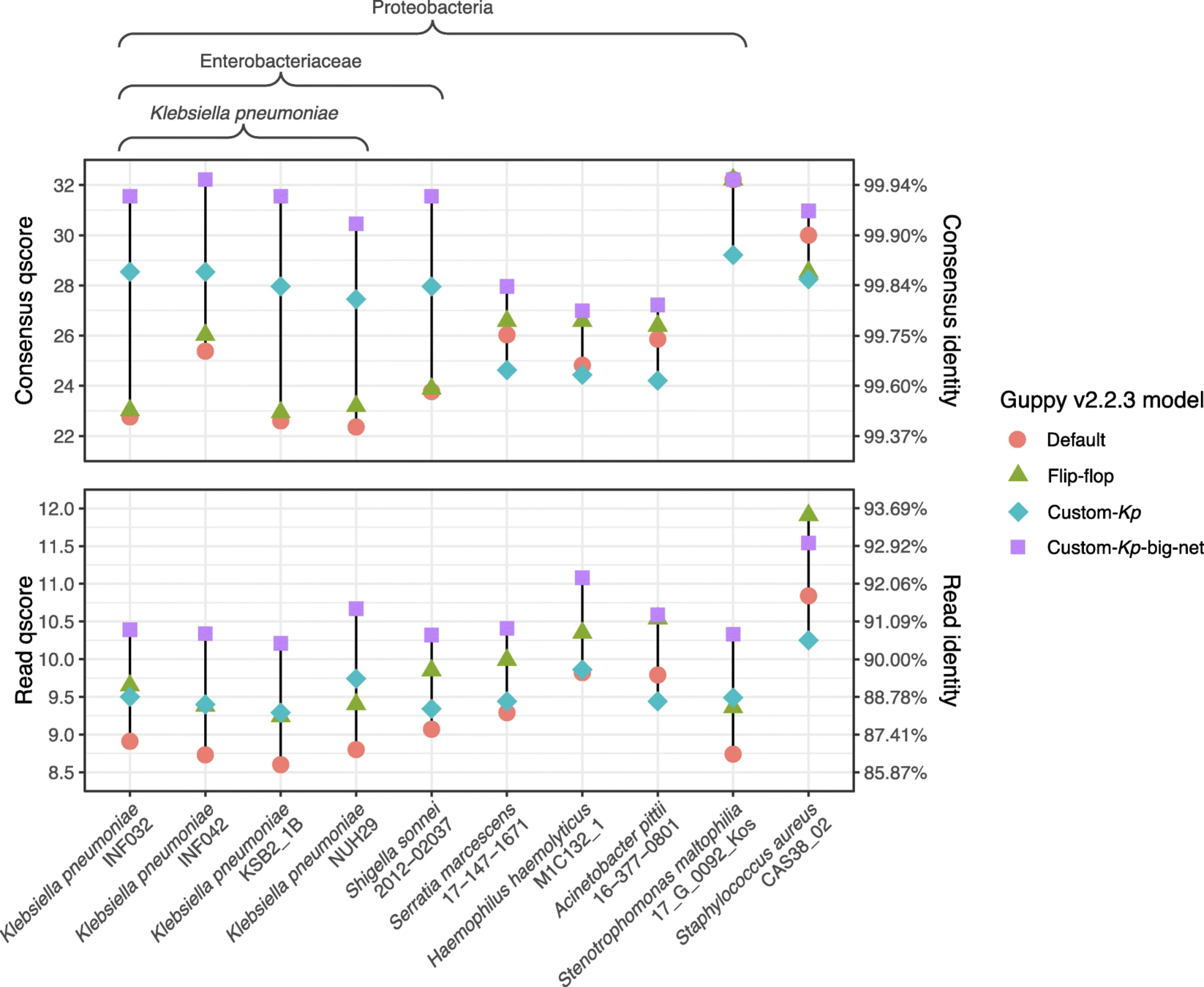
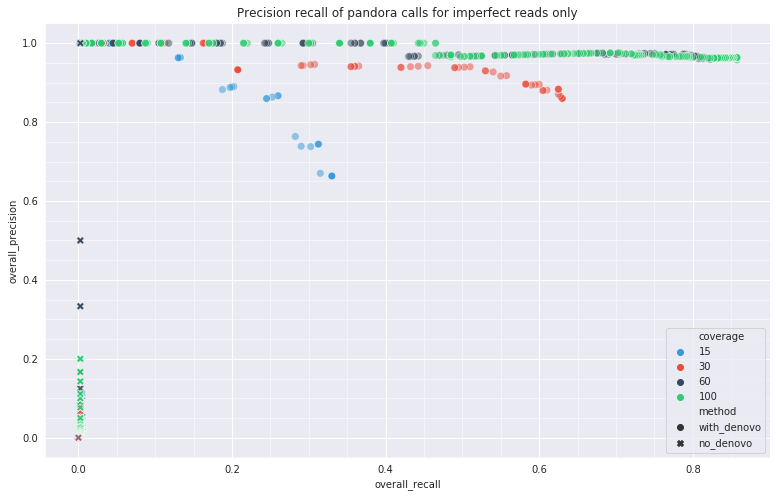
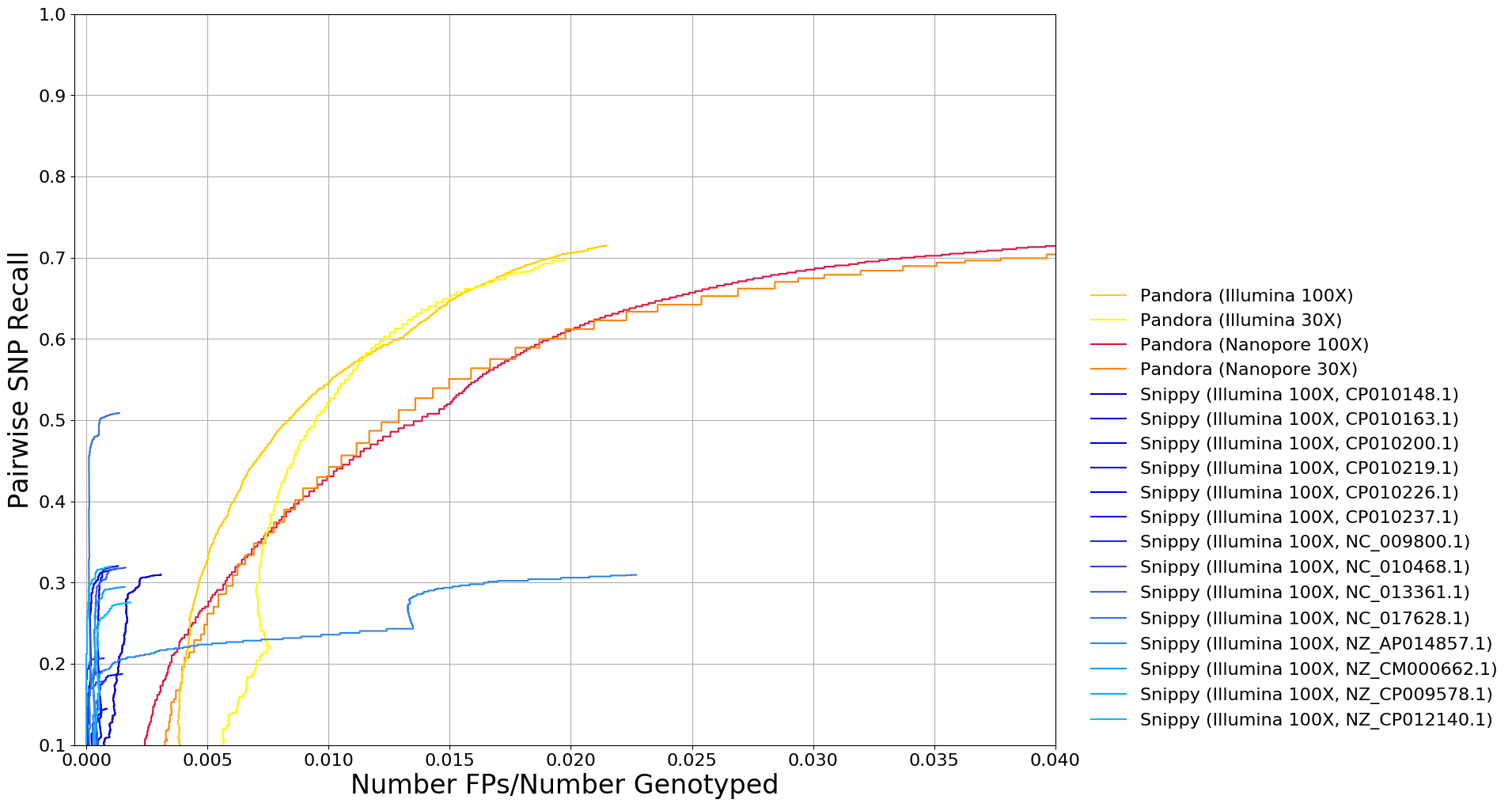
• Long Nanopore reads are noisy, and SNP calling with this data is not adequately benchmarked or standardised.
• Since Mycobacterium tuberculosis infects so many people, there is potential for considerable impact for clinical applications.
• There is also much to be gained from a high-quality pan-genome of M. tuberculosis as well as a detailed map of its enigmatic pe/ppe gene repertoire.
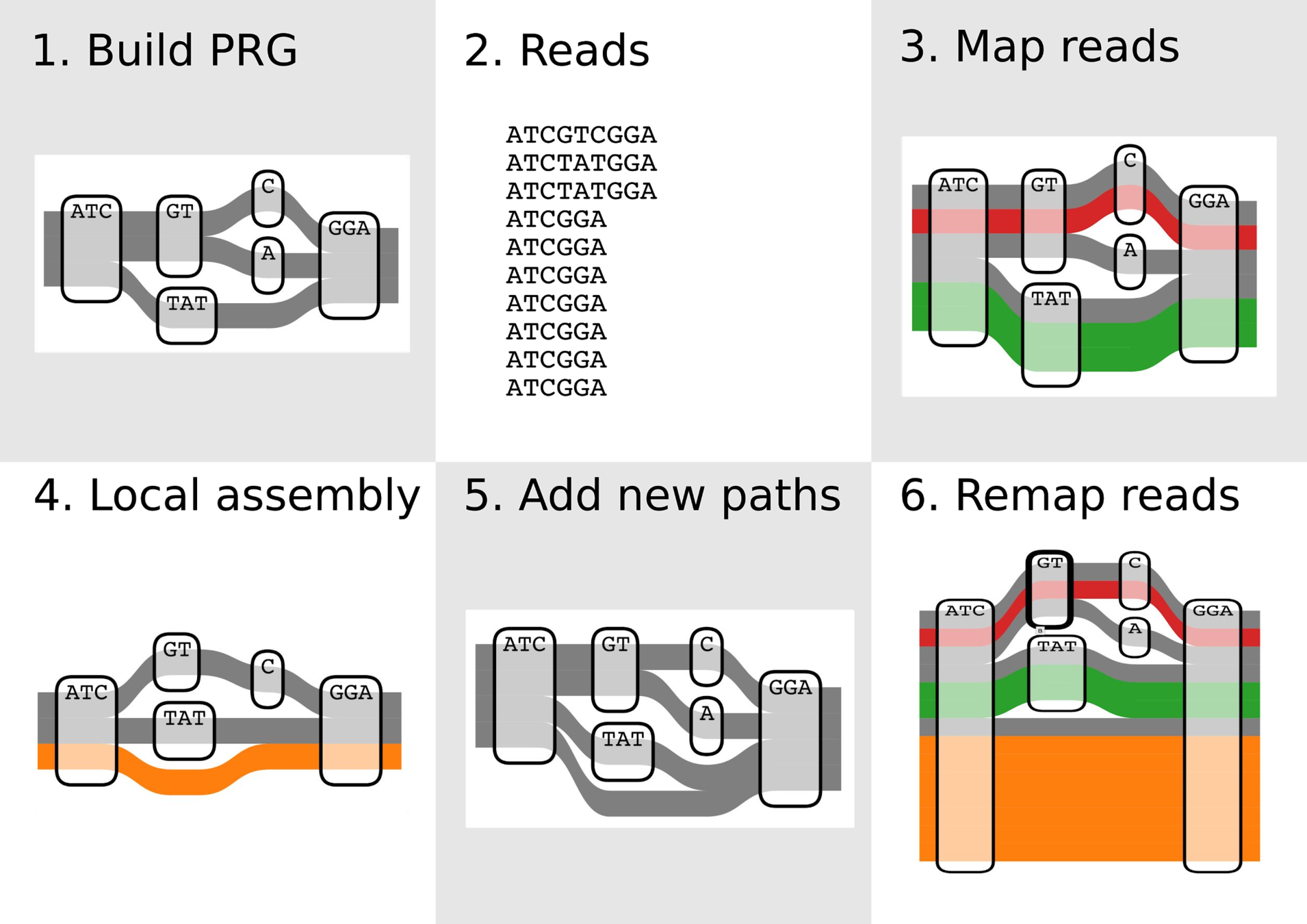
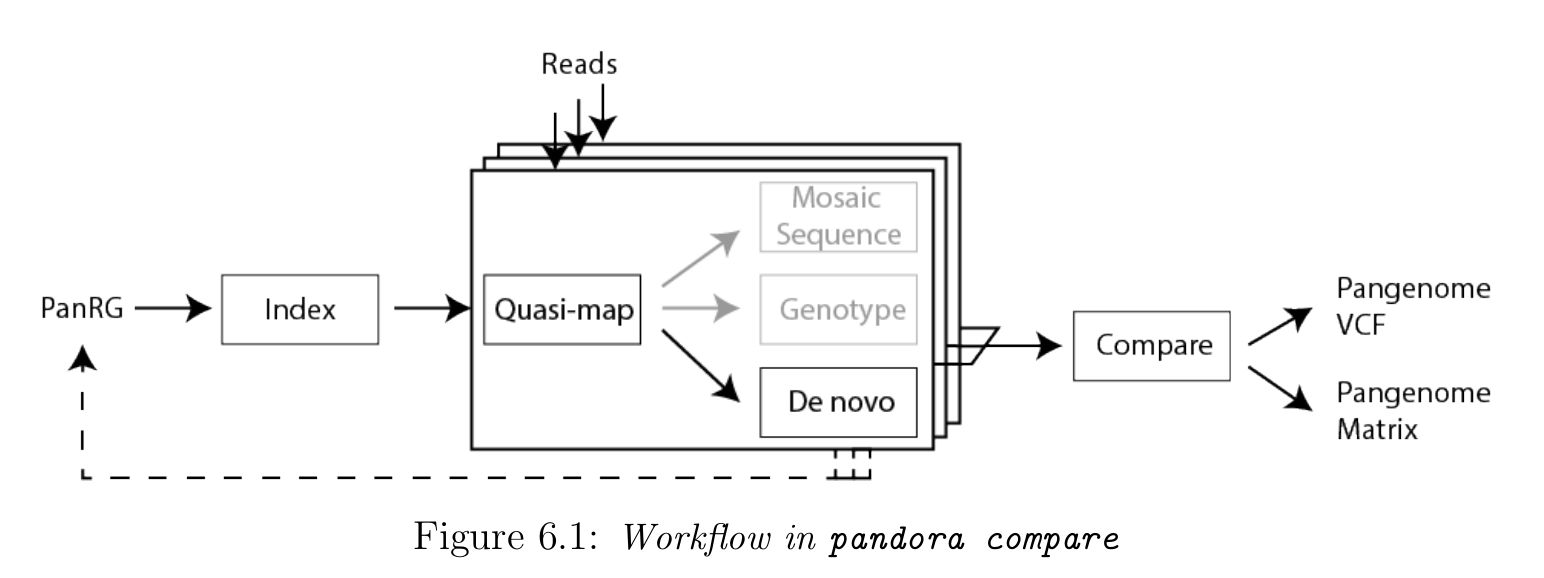
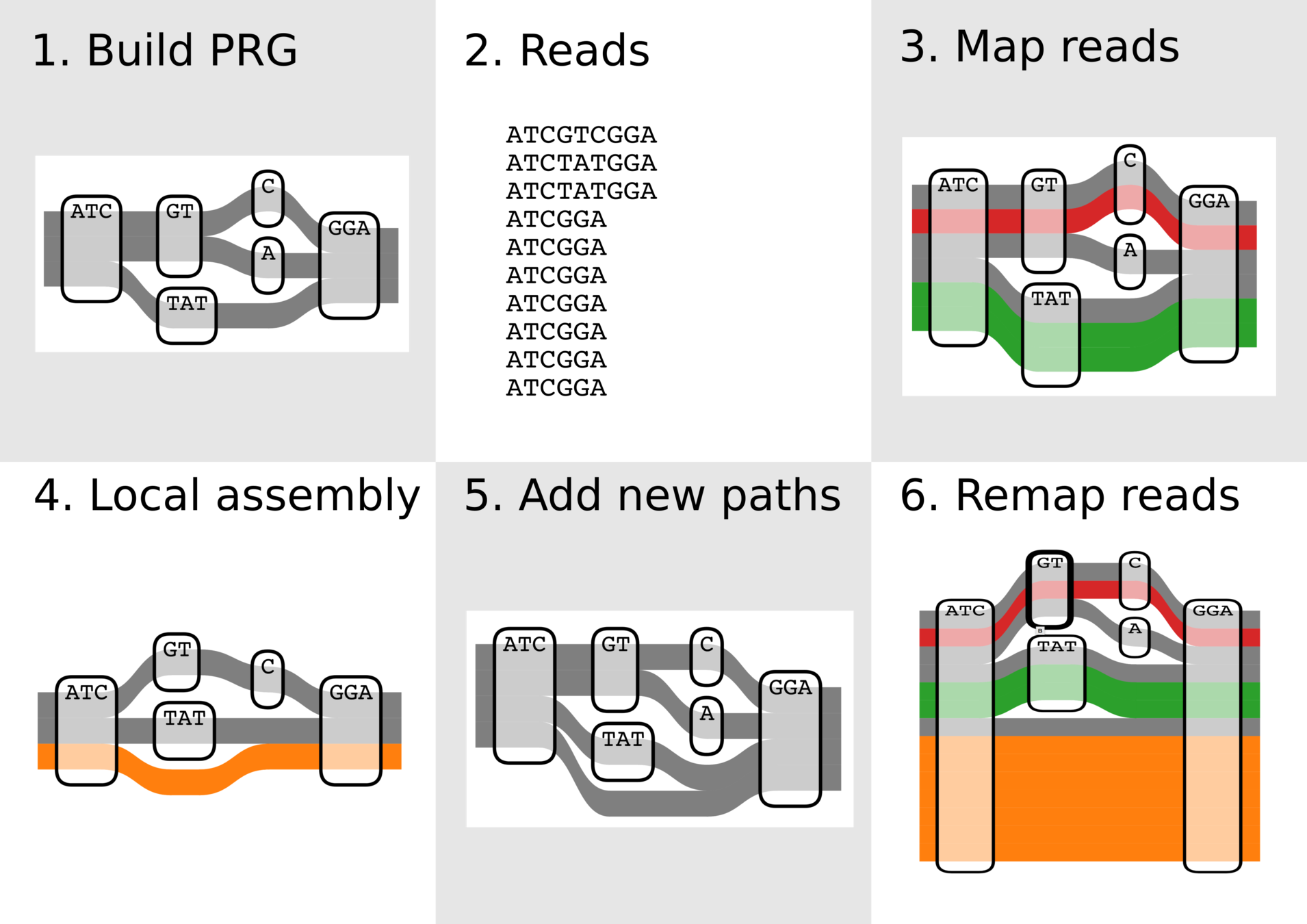
1. Develop algorithms and software for variant discovery using bacterial genome graphs, building on work of a previous student in the lab.
2. Benchmark Nanopore versus Illumina SNP calling, show our algorithms meet the needs of clinical and public health users, validate and publish.
3. Improve upon current whole-genome sequencing-based drug resistance prediction for M. tuberculosis using genome graphs.
4. Curate a high-quality reference pan-genome for M. tuberculosis that includes a detailed map of the pe/ppe genes.
Source: WHO
Source: WHO
Rachel Colquhoun
Rachel Colquhoun
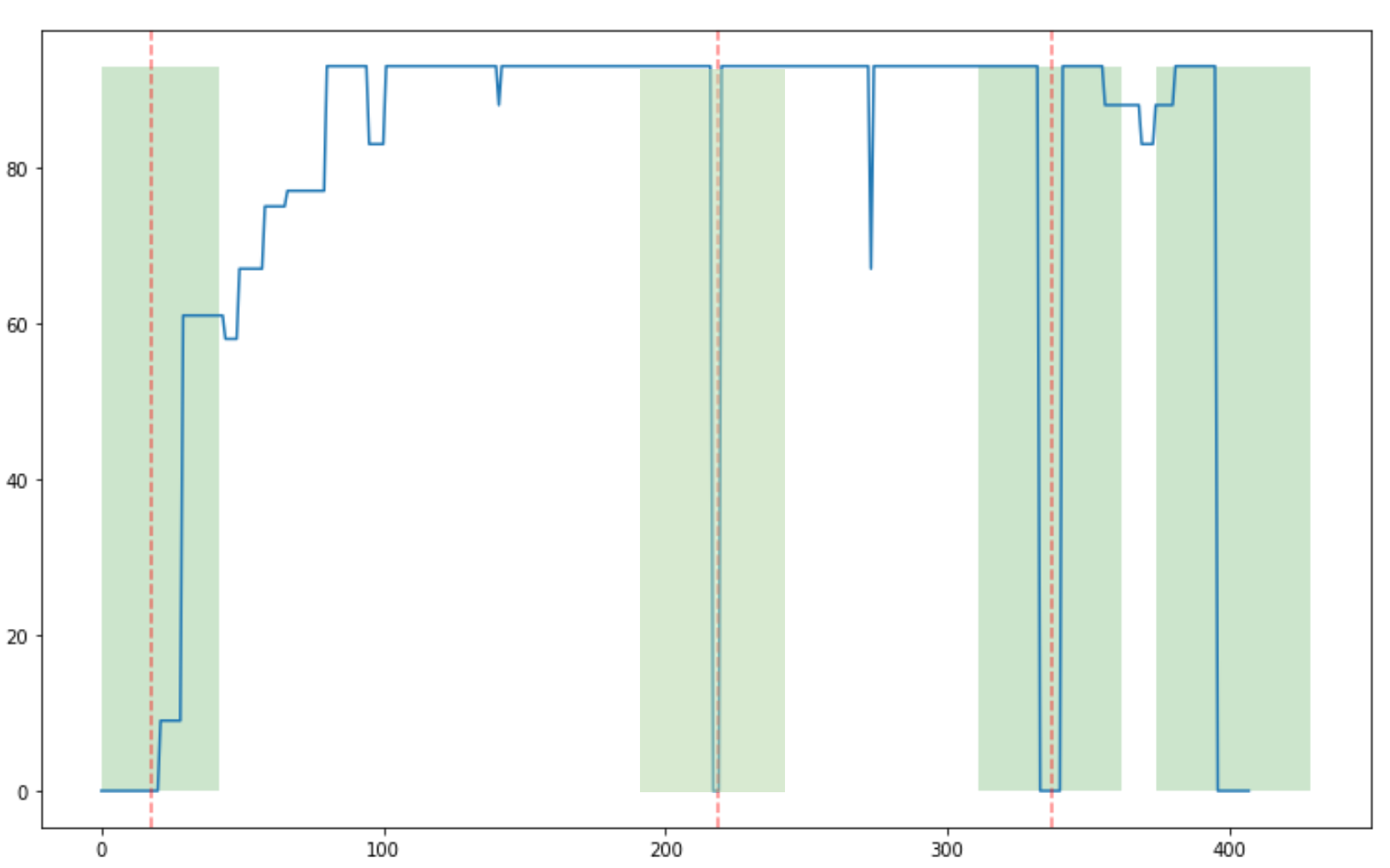
Per-base coverage
Position in gene (bp)
EMBL Heidelberg in February 2019
Co-taught with Josep Moscardó and organised by EMBL BioIT initiative
I taught and created material for making and running containers, how to share and store them, and also how to integrate into common workflow management systems.
bash more efficientlyEMBL-EBI in January 2019
Handy aliases I regularly use for working with sequencing data, a brief introduction to containers, searching their command history quickly, and advanced methods for finding files...
Most of 2018
London Calling - London, May 2019
By Michael Hall
Slide deck for my second-year presentation to my thesis advisory committee.



