Pedro Cerqueira
Lab Meeting 06/01/2020
Alignment-free approaches for sequence comparison: an overview
2. Alignment-free (AF) approaches
1. Alignment-based approaches
Summary
2.1 Definition
2.2 Categories of AF methods
3. Perfomance
4. Final remarks
1. Alignment-based approaches
- Phylogenomics is the study of evolutionary relationships via comparative analysis of genome-scale data, intersecting the fields of evolution and genomics.
- The first and key step for these studies is the comparative analysis of DNA and amino acid sequences. To achieve it, several programs were developed to perform sequence alignment.
- This method positions the biological sequences’ building blocks to identify regions of similarity that may have consequences for functional, structural, or evolutionary relationships.
1. Alignment-based approaches
Several alignment-based tools were created such as:
- Sequence similarity search tools (e.g. BLAST)
- Multiple sequence aligners (e.g. ClustalW, Muscle or MAFFT)
- Whole-genome aligners (e.g. progressive Mauve)
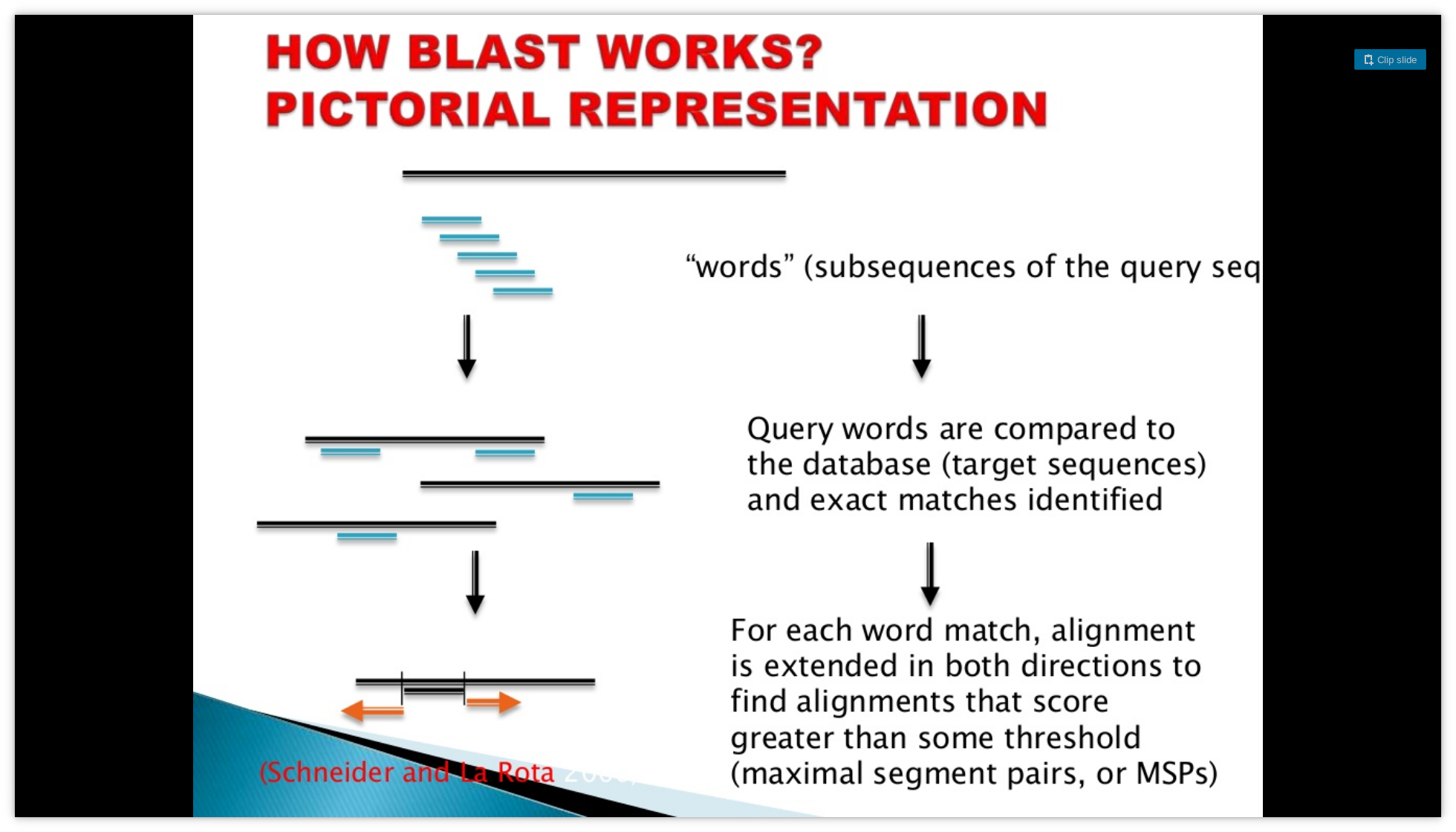
1.1 BLAST example
Zielezinski et al. (2017) have shown that in scenarios of low sequence identity these approaches are inaccurate.
Moreover, they assume that the linear order of homology is preserved within the compared sequences.
1. Alignment-based approaches
- Alignment-based programs assume that homologous sequences include a series of linearly arranged and more or less conserved sequence stretches (which is termed collinearity). For example, viral genomes exhibit great variation in the number of genetic elements.
- These approaches also depend on assumptions about the evolution of the sequences that are being compared. Small changes to input parameters can greatly affect the alignment.
- Finally, these approaches are memory and time consuming, where an accurate multiple sequence alignment cannot be solved in a realistic time frame.
2.1 Alignment-free approaches
- Any method of quantifying sequence similarity/dissimilarity that does not use or produce alignment at any step of algorithm application.
- Computationally less expensive;
- Resistant to shuffling and recombination events;
- Don't depend on assumptions regarding the evolutionary trajectories of sequence changes.
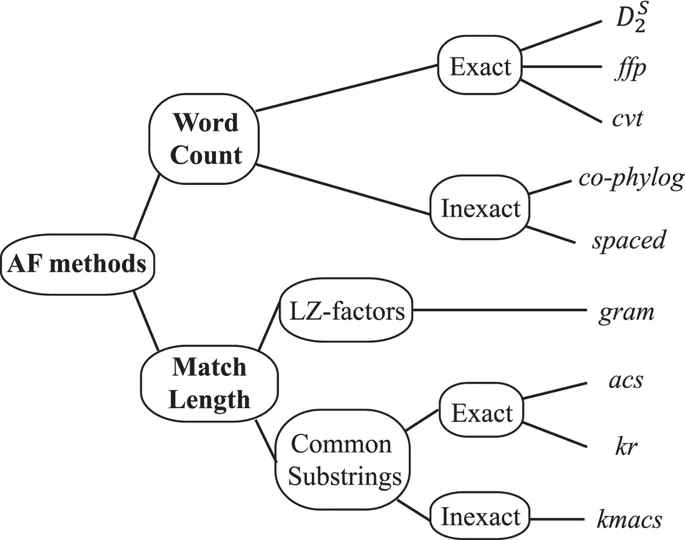
2.2 Categories of Alignment-free approaches
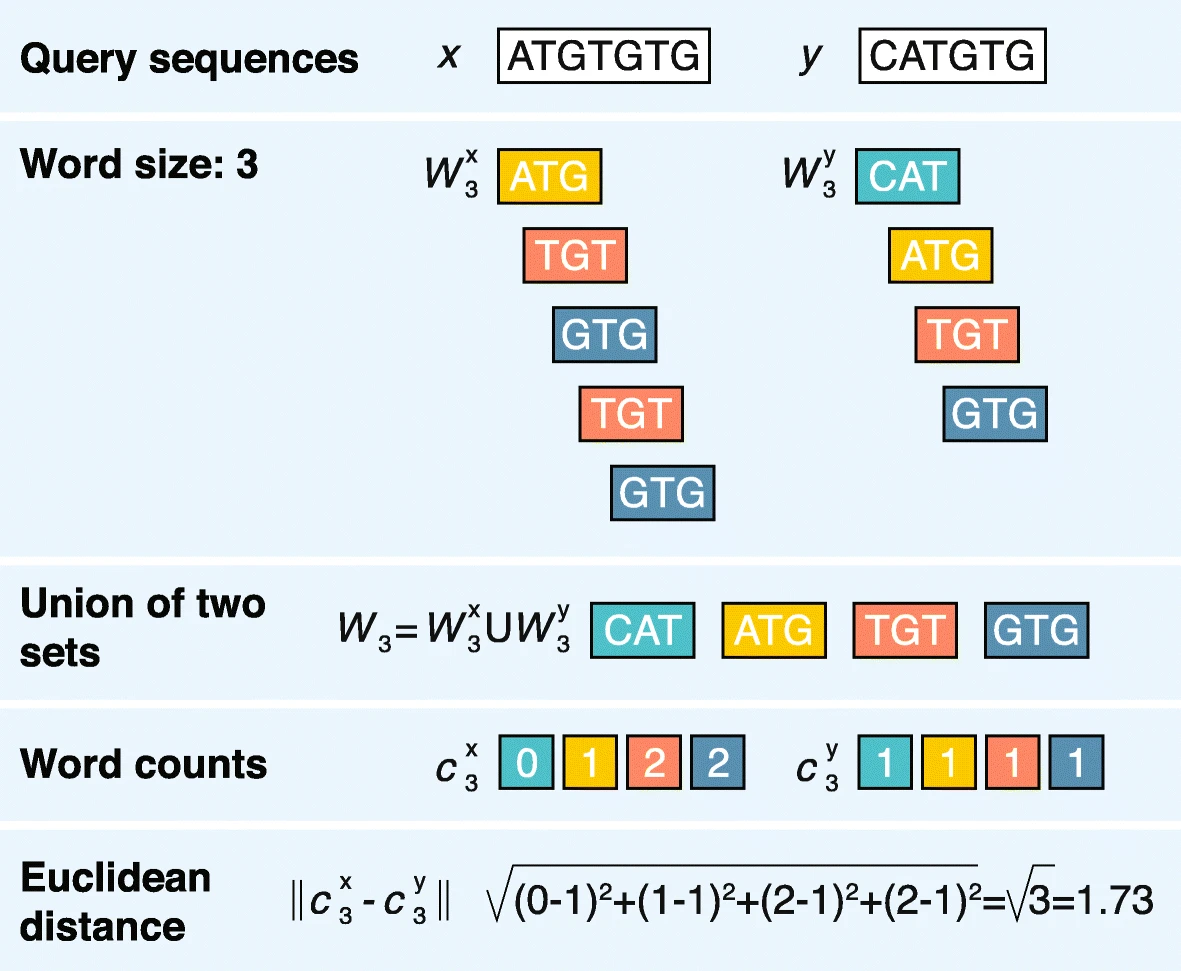
k-mers
2.2.1 Word-based methods
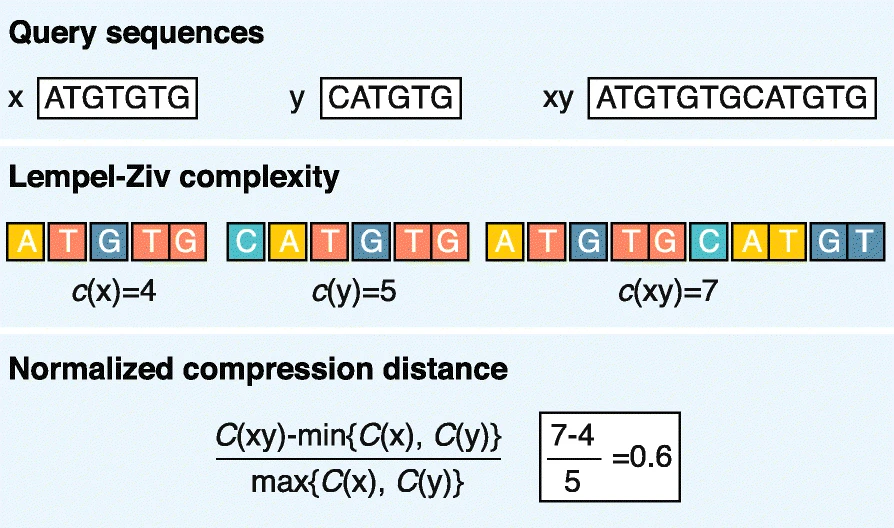
2.2.2 Match length methods
How are AF being used for NGS analysis?
-
Variant calling
- Traditionally mapping-based detection (GATK HaplotypeCaller and Samtools mpileup);
- AF methods allow for genotyping directly from NGS data, perfoming 1-2 orders or magnitude faster.
-
Taxonomic profiling
- Assignment of taxonomic labels wit near perfect accuracy (99%).
- CLARK, Kraken and Mash.
-
Assembly
- Correction of sequencing errors in raw reads.
3. How well do AF methods work?
- Vinga and Almeida (2004) and Hohl et al. (2007) showed that AF may perform better than traditional methods in case of proteins that underwent domain shuffling events;
- Dai and colleagues (2008) demonstrated that AF methods detected statistically relevant similarities in sequence compositions in contrast to traditional methods that showed only limited correspondence recognizable by alignments;
- Bernard and colleagues (2016) showed that AF methods:
- Most sensitive to the extent of sequence divergence;
- Less sensitive to low and moderate frequencies of horizontal gene transfer;
- Most robust against genome rearrangements.
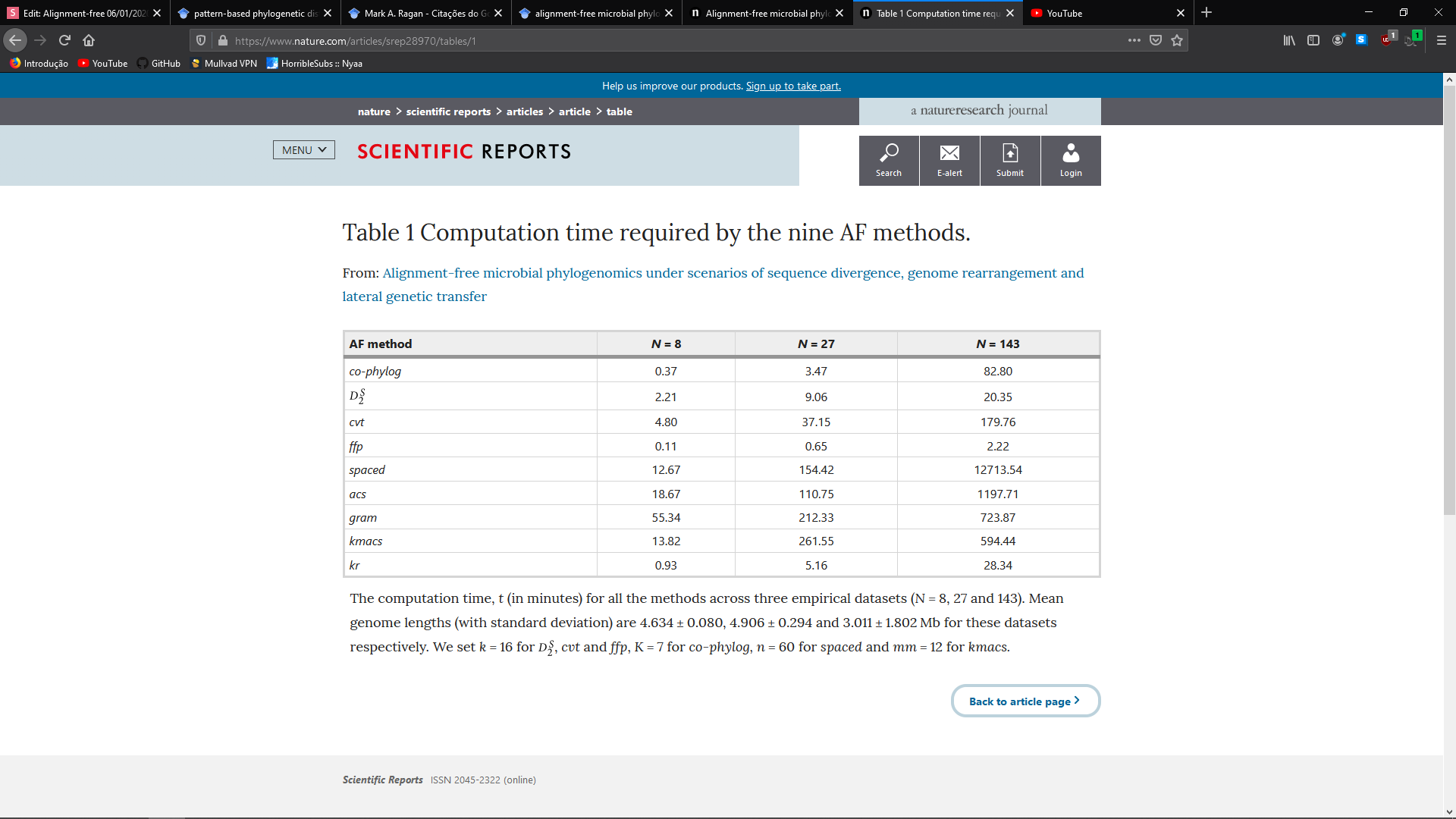
What is the optimal value of k?
3. How well do AF methods work?
4. Final remarks
- Should we just use AF methods instead of traditional methods?
- Not really.
- Alignments are still irreplaceable in many fields of biology such as annotation of conserved protein domains, reconstruction of ancestral DNA among others.
- AF are still young and many methods are theoretical and often evaluated with individually selected datasets.
Alignment-free 06/01/2020
By Pedro Cerqueira
Alignment-free 06/01/2020
Lab meeting




