Pharmacology 101
Swipe Left or Press Space
Our Pharm Project
First a Tutorial
Swipe Left or Press Right Arrow key

This is a Horizontal Slide
- Each horizontal slide talks about a New Drug
- To read more about a drug:
Swipe up or Press Down Key
This is a Vertical Slide
- Vertical slides will teach you more about each drug
- You can Swipe Left or Press Right Key to get the first drug
--------Or--------
- Swipe up or Press Down key to read more about the creators of this project
Saj Arora
- Role
- Creative Genius
- Improviser
- Team player
- Origins
- Toronto, Canada
- McMaster, Hamilton
- Yearbook Quote:
- Silly Rabbit, Tricks are for kids

Albert Cheng
- 2nd year NYUCD student
- Undergrad: SUNY Geneseo
- Hometown: New York
- Favorite sport: Basketball
- Favorite team: San Antonio Spurs
- Favorite Quote:

"Yesterday's the past, tomorrow's the future, but today is a gift. That's why it's called the present."
-Bil Keane
Steph Colaiacovo
Role
-
Disney enthusiast
-
Dreamer
-
Team player
Origins
-
Toronto, Canada
-
McMaster University, Hamilton
Yearbook Quote:
-
Hakuna Matata

Olga Degtyareva
Role
-
Candy Crush aficionado
-
Excels at vertical tasks
-
Team player
Origins
-
Priozersk, Russia
-
River Edge, NJ
“Hey Olga, are you still Russian?”
- “Yes.”
“Then why don’t you slow down??”

Dan Kim
Role
-
Jack of all trades
-
Automotive technician
-
Mechanical engineer
-
Father of two
Origins
-
Temple University
-
Philadelphia, Pennsylvania
Yearbook quote
-
“Imagination is more important than knowledge”

Alex Pisapia
Role:
- Team Motivator
Undergrad:
- Syracuse University class of 2012,
Origins:
- Staten Island New York
Quote:
- "Do or do not, there is no try"

Brendan Ruby
Nickname: Rubes
Origins:
- Madison, CT University of Connecticut, 2012
Favorite Hobbies:
- Knitting yarn
- Dress up
Quote:
- "Scared money don't make money"

Herbal Remedies
Brendan Ruby

Aloe Vera
Herbal Remedy
I drink Aloe Vera all the time
Mechanism of Action
- Acemannan, a complex carbohydrate isolated from Aloe leaves, has been shown to accelerate wound healing and reduce radiation induced skin reactions.
-
The mechanism of action of acemannan appears to be twofold.
- First, acemannan is a potent macrophage-activating agent and therefore may stimulate the release of fibrogenic cytokines.
- Second, growth factors may directly bind to acemannan, promoting their stability and prolonging their stimulation of granulation tissue.
Pharmacokinetics
- Aloe vera gel directly stimulates the activity of macrophages and fibroblasts.
-
Mannose 6-phosphate, the principal sugar component of Aloe vera gel, may be partly responsible for the wound healing properties of the gel.
- Binds to the growth factor receptors on the surface of the fibroblasts and thereby enhance their activity.
Drug Interactions (mechanisms of interaction)
- Cathartic effect of latex form often hastens passage of oral medications, often inhibiting their absorption, and may potentiate anticoagulant therapy by reducing intestinal absorption of vitamin K.
Drugs that might interact with them
- Some medicines MAY interact with aloe vera gel. However, no specific interactions with aloe vera gel are known at this time. Do NOT use Aloe vera if you are allergic to any ingredient in aloe vera gel.
Additional Information
- Used in mouth rinse, toothpaste, lubricating gel, antiseptic gel
- Possible uses as an anti-inflammatory, antiseptic, as well as promoting healing of canker sores and wounds
Asian ginseng
Herbal Remedy
Mechanism of Action
- Works as an adaptogen and immunomodulator
- Important substances: ginsenosides or panaxosides
- Stimulate the proliferation of hepatic ribosomes, increase natural killer-cell activity, and possibly enhance the production of interferons
Pharmacokinetics
- Absorption rate of ginseng saponins is low after oral administration
- Extensive metabolism in the gastrointestinal tract, poor membrane permeability, and low solubility of deglycosylated products limit intestinal absorption of ginseng saponins
- High oral doses may saturate metabolism and increase bioavailability.
- Liver and bile clear ginseng saponins from circulation
- Cytochrome P450 catalyzed ginsenoside metabolism, and it has been described that CYP3A4 catalyzed metabolism by oxygenation the hepatic disposition of ginsenosides
Drug Interactions (mechanisms of interaction)
- Inhibited blood clotting from effects on platelet adhesion and blood coagulation
- “Ginseng abuse syndrome,” with diarrhea, hypertension, and nervousness, has been described
- Various interactions with drugs listed on next slide
Drugs that might interact with them
- Heart Medications
- Blood thinners
- Caffeine
- Psychiatric medications
- Morphine.
Kava-Kava
Herbal Remedy
Mechanisms of Action
- The mechanism of action of kava has not been fully elucidated, but multiple effector sites are involved.
- Anxiolytic and sedative effects of kava suggest that it potentiates γ-aminobutyric acid (GABA) inhibitory neurotransmission.
- Kavalactones and pentobarbital also produced a synergistic effect on [³H] muscimol binding to GABA.
- Kavalactones inhibit voltage-dependent sodium and calcium channels
- May exert its effects through neurotransmitters such as dopamine and serotonin
Pharmacokinetics
- Kavalactones have a half-life of 9 hours and achieve peak plasma levels 1.8 hours after administration
- In rats, dihydrokawain was completely excreted within 48 hours, primarily through urinary excretion of hydroxylated metabolites
- Bile and feces did not appear to be important routes of excretion
Drug Interactions (mechanisms of interaction)
- Summation of effects with benzodiazepines and other CNS depressants.
- High doses may increase dystonic reactions with antipsychotics and levodopa.
Drugs that might interact with them
- Alcohol
- Anxiolytics
- Dopamine-agonists
Additional Information
- Kavalactones, also known as kavapyrones or -pyrones, are responsible for most of the pharmacological effects.
-
The six major kavalactones that have been identified:
- kawain, dihydrokawain, methysticin, dihydromethysticin, yangomin, and desmethoxyyangonin
- Local anesthetic action causes temporary mouth numbness
- May rarely cause hepatotoxicity and liver failure
- High doses may cause inebriation, with incoordination, ataxia, and drowsiness
St. John’s Wort ( Hypericum perforatum )
Herbal Remedy
Mechanisms of Action
- St. John's wort is a botanical remedy with a long history of use for depression.
- Many biologically active components, including hypericin, hyperforin, and some flavinoids.
- Blocks the reuptake of 5-HT, NE, dopamine, GABA, and glycine with approximately equal potency, a unique therapeutic property.
- Hyperforin may reduce the Na+ gradient on which the symporters depend, decreasing neurotransmitter uptake.
- St. John's wort can also block MAO-A and MAO-B at high doses
Pharmacokinetics
- Very effective inducer of hepatic CYPs, specifically, CYP3A4, CYP2C9, and CYP1A2
- Induction of CYPs leads to significant decreases in plasma levels of drugs that are substrates of the induced CYPs
- Oral ingestion: plasma levels were measurable within two to three hours.
- The elimination half-life was 24 to 48 hours.
Drug Interactions (mechanisms of interaction)
- Shown to cause multiple drug interactions through induction of the cytochrome P450 enzymes CYP3A4 and CYP2C9 , and CYP1A2
- Phototoxic/photoallergic reactions with tetracyclines, sulfonamides, and proton pump inhibitors
- Summation effects with benzodiazepines, opioids, and other CNS depressants
- Serotonergic crisis possible with meperidine, MAO inhibitors, and other antidepressants
- Decreases plasma concentrations of protease inhibitors, cyclosporine, digoxin, and warfarin
Drugs that might interact with them
- Tetracyclines
- Sulfonamides
- Proton pump inhibitors
- Benzodiazepines
- Opioids
- CNS depressants
- Meperidine
- MAO inhibitors
- Other antidepressants
- Protease inhibitors
- Cyclosporine
- Digoxin
- Warfarin
Antibiotics
Saj Arora

Amoxicillin
Antibiotics
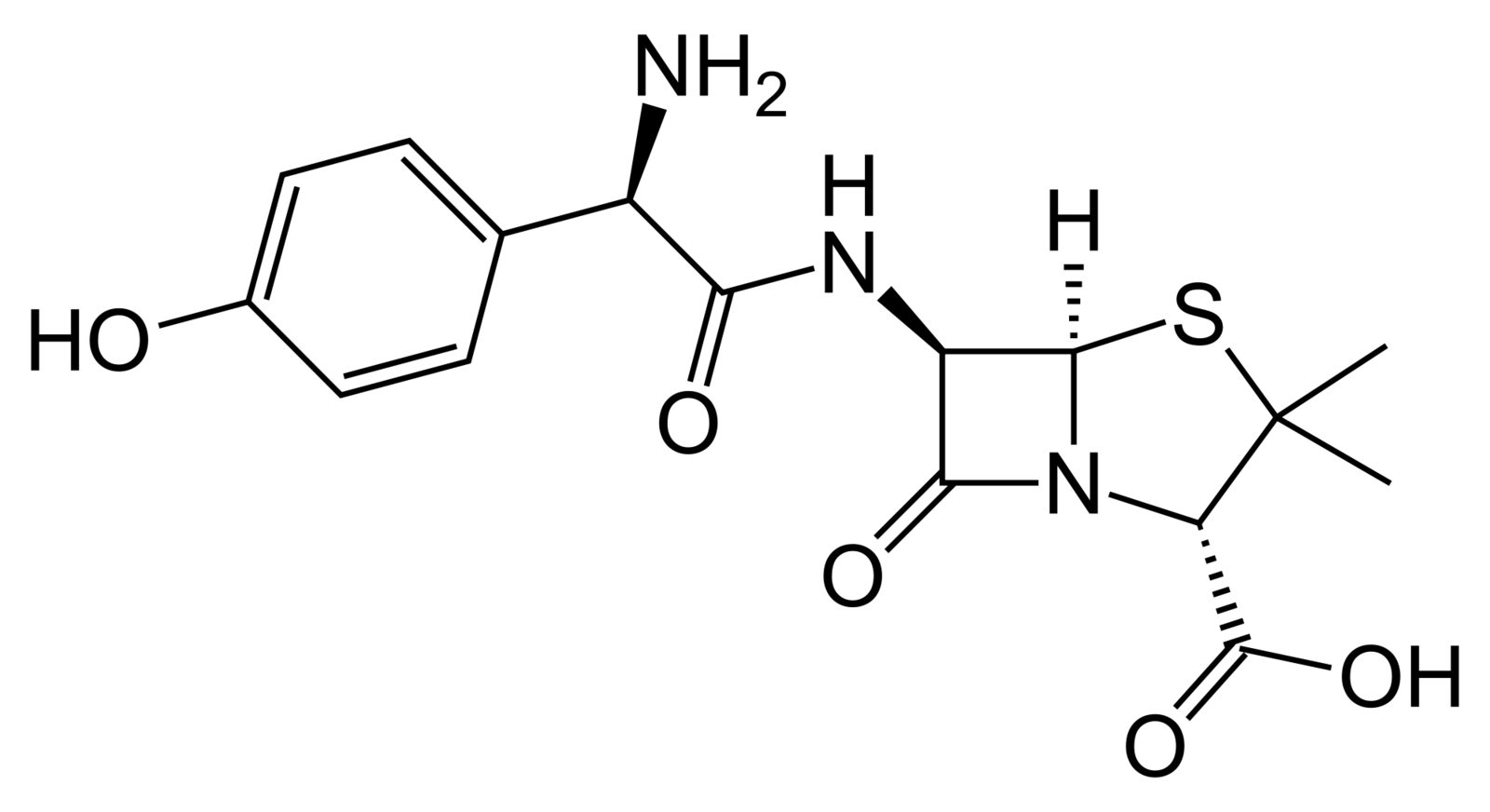
Mechanism of Action
- Amoxicillin is an effective bacteriostatic agent
- Amoxicillin is a penicillin-like antibiotic
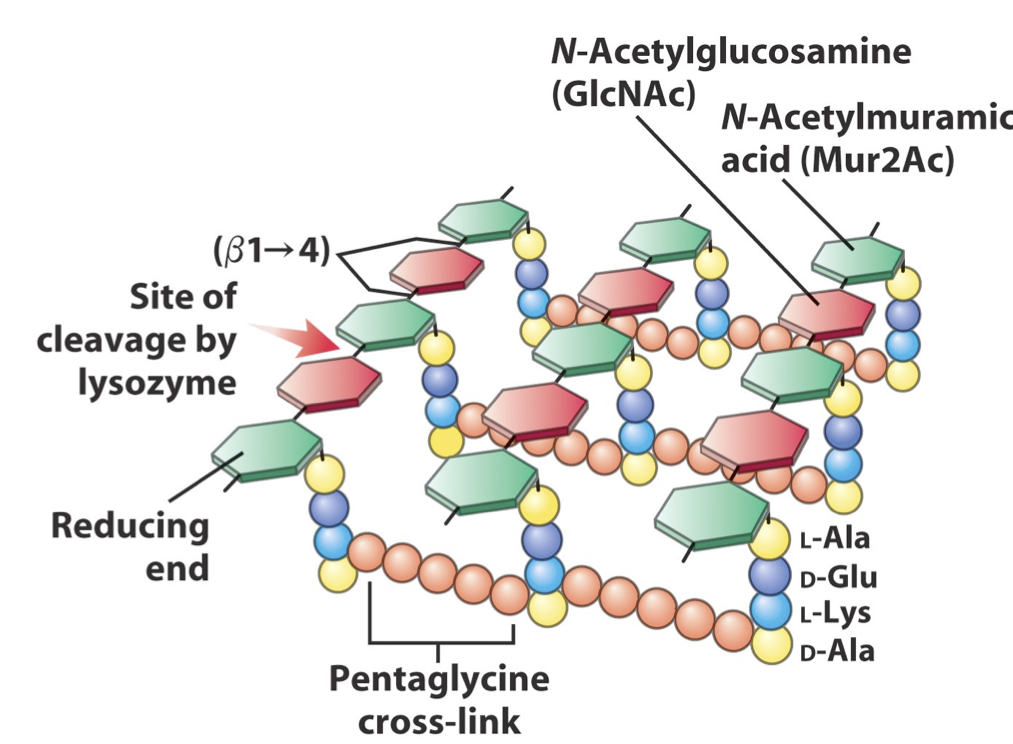
- Bacterial walls are synthesized of repeating monomers of peptidoglycan
- Amoxicillin inhibits cross-linkage between peptidoglycan chains by inhibiting the beta-lactam enzyme
- Beta-lactamase decrease the effectiveness of this drug
Pharmacokinetics
Absorption
- Readily absorbed as it stays intact in gastric acid
- Peaks at 1 to 2 hours after administration
Distribution
- Reaches most areas of the body except for the brain and spinal fluid
- 20% of Amoxicillin is bound to plasma protein
- Amount of plasma protein regulates activity.
Pharmacokinetics
Metabolism and Excretion
- Half life is 61.3 minutes
- 60% of orally administered amoxicillin is excreted in urine within 6 to 8 hours.
- serum levels are observed up to 8 hours after an orall does
Drug Interactions
- Pencillin based drugs like Amoxicillin can decrease renal clearance of other drugs
- Methotrexate is heavily cleared by the kideny
- Methotrexate build up leads to nausea, vomiting, mouth ulcers and lower blood cell count
- Translates to increased chances of infections and bleeding problems.
- Amoxicillin can interact with other antibiotics and its effect can be synergistic (e.g. Erythromycin, Azithromycin)
- Has been used in clinical setting for dealing with H. Pylori infections.
Additional Information
Method of Resistance
-
Bacteria can s ynthesize beta-lactamases to cleave the beta-lactam ring of amoxicillin
-
Renders drug useless as it can't bind the enzyme
H. pylori Eradication
- H. Pylori infections studied in a randomized, clinical trial
- Triple therapy and possible dual therapies that include Amoxicillin were shown to be effective in lowered H. Pylori in patient
- Significant reduction in the risk of duodenal ulcer recurrence.
Clindamycin
Antibiotics
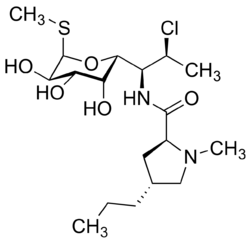

Mechanism of Action
- Clindamycin works on the bacterial ribosome
- Ribosomes are the protein production factory in cell
- Without proteins the bacteria can not function
- Clindamycin works by inhibiting the 50S subunit of the ribosomes
- Clindamycin is effective in dealing with both Gram positive and Gram negative infections
- It is bacteriostatic so it inhibits growth of bacteria.
Pharmacokinetics
Absorption (oral)
- Clindamycin is rapidly absorbed and peaks in serum at 45 minutes with absorption around 90%
- Food is not a modifier and no dosage alteration is necessary for the elderly with normal hepatic function and normal (age adjusted) renal function.
Distribution
- Clindamycin is widely distributed through all body fluids and tissues
- Found in bone tissue and has a biological half life of approximately 2.4 hours
- Clindamycin is not usually found in CSF so it doesn’t cross the blood brain barrier readily.
Pharmacokinetics
Metabolism and Excretion
- From the bioactive agent in circulation, 10% of Clindamycin is excreted in urine and 3.6% in feces
- The remainder of the clindamycin is excreted as biologically inactive metabolites.
- Serum half-life of clindamycin is increased in patients with renal problems and those on hemodialysis.
Drug Interactions
- Lincomycin and erythromycin exhibit an antagonistic relation with clindamycin
- They both work on the ribosomes, binding to the 50S subunit
- Decrease the effectiveness of the other drug due to competition for the same site.
- Clindamycin has neuromuscular blocking properties and interactions with other neuromuscular blocking agents such as curare can be significant.
Drug Interactions
-
Clindamycin is a broad spectrum which can decrease the flora in the intestine and can significantly alter the enterohepatic cycling of certain drugs
-
Ethinyl estradiol, like in the birth control pill, would have decreased effectiveness in patients on clindamycin
-
Verapmil is a hypertension drug whose effect is shown to increase with clindamycin usage
-
Cyclosporine is an immunosuppresant and its effect is decreased with clindamycin
-
Additionally, d ue to destruction of gut bacteria, diarrhea and watery stool is a common problem in patients on clindamycin.
Azithromycin
Antibiotics
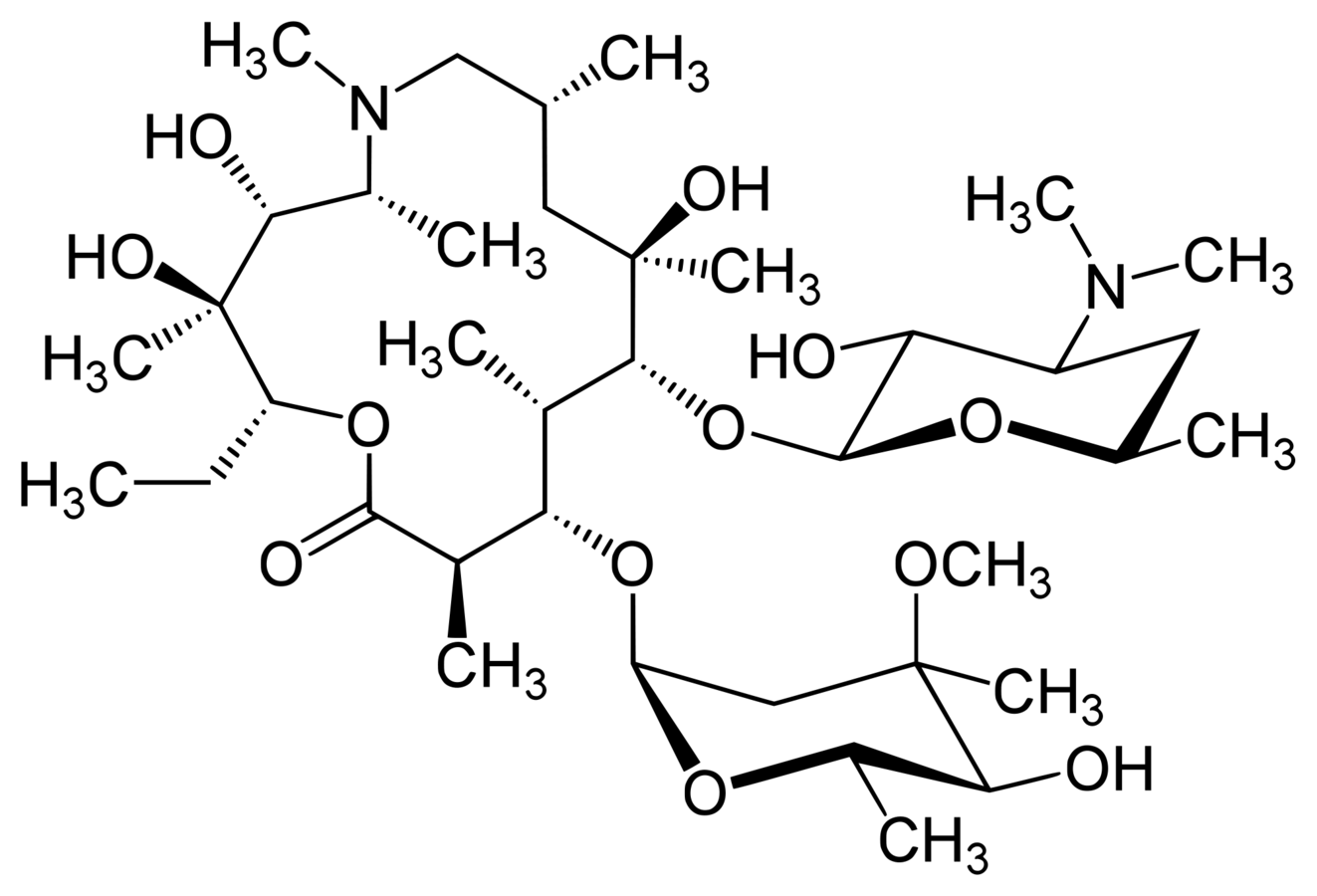
Mechanism of Action
- Azithromycin, an erythromycin derivative, is a macrolide antibacterial
- Macrolide antibiotics inhibit protein synthesis
- Azithromycin inhibits peptidyltransferase, a critical enzyme in the transferring of amino acids during protein synthesis
- Elongation of the amino acid chain is stopped and protein synthesis comes to a halt
- Bacteria require protein synthesis to perform most functions and for growing
- Hence, macrolide antibiotics are bacteriostatic.
Pharmacokinetics
Absorption (oral)
- Azithromycin can be taken in both a tablet form and as an oral suspension
- After first-pass metabolism the bioavailability of the drug drops down to about 34%.
- Absorption of the drug increased insignificantly when taken with food.
Distribution
- Albumin is the largest proponent of the plasma proteins
- Free drug concentration can be variable depending on the level of protein binding.
Pharmacokinetics
Distribution (cont'd)
- Therefore, effective concentrations can range from 51% to 7% at 2 μg/mL.
- Azithromycin penetrates into the skin, lung, tonsil, and cervix.
- Additionally, it is also found in bone, prostate and gallbladder.
Metabolism and Excretion
- Azithromycin has an average half-life of 68 hours.
- It is predominantly clearer by the liver via biliary excretion as an unchanged drug.
- Approximately 6% is renally cleared.
Drug Interactions
- CNS toxicity is a concern with the use of azithromycin and anti-psychotic medication such as Pizomide and Clozapine.
- Azithromycin decreases the rate of metabolism of Pizomide leading to increased duration and activity of the drug.
- This can increase the risk of abnormal heartbeat (prolonged QTc interval).
- On the other hand, the mechanism for the interaction between Clozapine and azithromycin is not as well understood.
Drug Interactions
-
Other than pizomide, methadone and other other CNS altering drugs should be prescribed with diligence as azithromycin has the potential of altering rates of metabolism and excretion.
Additional Information
-
Azithromycin is a broad spectrum antibiotic, hence it has the capacity to wipe out much of the gut flora leading to watery diarrhea and watery stool.
-
This can lead to dehydration as well as loss of electrolytes which can cause ion deficiencies and lead to nausea and vomiting.
Antifungals
Albert Cheng
Nystatin
Antifungal
Description
- Polyene antibiotic derived from Streptomyces noursei
- Used mostly in topical applications to clear fungal infection of the mucosa, skin, and vagina
-
Comparison to Amphotericin B
- Similar structure
- Slightly smaller spectrum

Mechanism of Action
- Similar MoA to Amphotericin B
- Forms a complex with ergosterol - a major component of the fungal cell membrane
- Complexes form pores in the membrane that leads to K leakage
- This alters cell membrane permeability and leakage of intracellular component
- Ultimately, results in cell death
Pharmacokinetics
- Not well-absorbed through skin, mucous membranes, or GI tract, thus, very little metabolism
- NEVER given parenterally because of systemic toxicity!
- Orally ingested Nystatin has 0% bioavailability and passes unchanged through GI tract
- Elimination occurs via feces
- Renal insufficiency patient - Nystatin can accumulate in the plasma
Adverse effects
- Main problem - nasty bitter, foul taste
- Also diarrhea, nausea, and vomiting
- Oral sensitization and irritation may occur
- Rarely, rashes, tachycardia, bronchospasm, facial swelling and muscles are reported
Additional Information
- NO KNOWN DRUG INTERACTION with Nystatin
- Relatively insoluble in water and unstable except as a dry powder
- Oral Nystatin should only be given to pregnant women when clearly needed
- Should not be used to treat systemic fungal infections
- Effective against many species of:
- Candida, Histoplasma, Cryptococcus, Blastomyces and dermatophytes ( Epidermophyton, Trichophyton, Microsporum)
Implications in Dentistry
- Topical Nystatin is the drug of choice for treatment of Candidal infections of the oral cavity
- Oral Moniliasis
- Thrush
- Denture stomatitis
- To treat oral candidiasis
- 2-3mL of a suspension is placed in the mouth, swished, and held for at least 5 minutes before swallowing
- To treat denture stomatitis
- Apply Nystatin ointment topically every 6 hours
Caspofungin
Antifungal (Class: Echinocandins)
Description
- Derived from fermented by-product of Glarea lozoyenisi
- 1st Echinocandin approved by FDA for clinical use
- Main use:
- Treatment of febrile neutropenia
- Prophylaxis against Candida infections in hematopoietic stem cell transplant patients
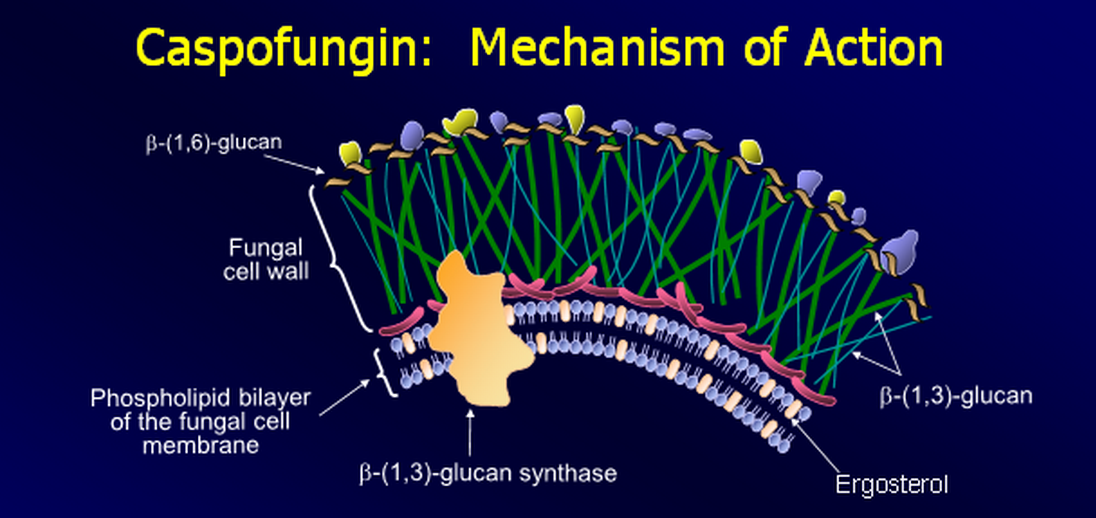
Mechanism of Action
- Non-competitively inhibition of 1-3-beta glucan synthase
- Inhibits the synthesis of beta (1-3)-D-glucan
- Beta (1-3)-D-glucan synthesis does not occur in human cells
- Loss of beta-glucan results in loss of cell wall integrity
- This results in lysis of the fungal cell due to inability to resist osmotic forces
Pharmacokinetics
- High molecular weight, thus they are poorly absorbed via oral ingestion (not active)
- High affinity for serum proteins
- Half-life: 9-11 hours
- Slowly metabolized via hydrolysis and N-acetylation
- Drug elimination is approximately equal between the urine and feces
- NO primary interactions with CYP450 or P-glycoprotein pumps
Drug Interactions
- Cyclosporine
- Carbamazepine
- Dexamethasone
- Efavirenz
- Nevirapine
- Phenytoin
- Rifampin
- Tacrolimus
Drug Interactions
- Cyclosporine can increase plasma levels of Caspofungin and leads to increase risk of liver disease
- Enzyme inducers may decrease the effectiveness of Caspofungin
- Caspofungin reduces the plasma levels of Tacrolimus and its efficacy
- Dose adjustments need to be considered with the drug interations mentioned above
Additional Information
- Combination therapy with other antifungal agents has synergistic effects against cryptococcal species
- Higher therapeutic efficacy against Candidal infections compared with Amphotericin B in immunocompromised patients
- Important in patients with life-threatening systemic fungal infection who cannot tolerate Amphotericin B or Azoles
-
Adverse effects:
- Rash, facial swelling, pruritis, or sensation of warmth
Fluconazole
Antifungal (Class: Azoles)

Description
- Water-insoluble, fluorine-substituted bistriazole
- Effective antifungal in immunocompromised patient
- Drug of choice for:
- Cryptococcus neoformans
- Coccidioidomycosis
- Candidemia
- Administered orally or IV
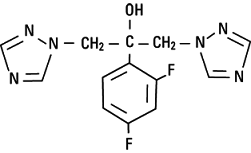

Mechanism of Action
- Inhibits the fungal cytochrome P450 enzyme 14-alpha-demethylase
- Enzyme converts lanosterol to ergosterol
- Drug blocks the demethylation of lanosterol to ergosterol
- This results in the accumulation of 14-alpha-methyl sterols in fungi and confers its fungistatic activity
Pharmacokinetics
- Absorption is excellent whether orally or IV
- Minimal affinity for serum proteins
- Bioavailability is not affected by the absence of stomach acid
- Half-life: 30 hours
- Elimination occurs via renal excretion
- 80% of administered oral doses appear as the unchanged drug in urine
- Reduced dose in patients w/ compromised renal function
- 10% of elimination is due to metabolism
Drug Interactions
- Berberine
- Coumadin-type anticoagulants
- Phenytoin
- Cyclosporine
- Rifampin
- Azithromycin
- Cisapride
- Astemizole
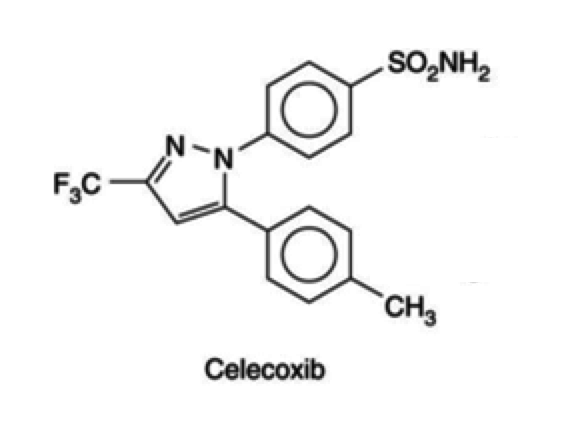
- Celecoxib
- Voriconazole
- Tacrolimus
- Tacrolimus
- Short-acting BZD
- Tofacitinib
- Triazolam
- Oral Contraceptives
- Carbamazepine
- Quinidine
- Hydrochlorothiazide
- Amphotericin B
- Calcium Channel Blocker
- NSAIDs
Drug Interactions
- Inhibits the human cytochrome P450 enzyme
- Particularly (CYP2C19 and CYP3A4/CYP2CP)
- This decreases metabolism of other drugs and increases bioavailability
- Can prolong QT interval and increase risk of cardiac arrhythmia
- Berberine has some synergistic effects with fluconazole in drug-resistant C. albicans infections
- May decrease the metabolim of Benzodiazepines
Implications in Dentistry
- Effective for treatmet of mucosal candidiasis, including oropharyngeal and esophageal candidiasis
- Weekly use of fluconazole provides prophylactic value against mucosal candidiasis in HIV patients
- Also used in primary treatment of coccidioidal meningitis and blastomycosis and histoplasmosis
- Fluconazole is more effective against oral candidiasis that Nystatin in immunocompromised children
Additional Information
- LACK endocrine side effects seen with Ketoconazole
- Excellent permeability into CSF of normal/inflamed meninges
- Fluconazole along with other Azoles should not be used in pregnant patients because it is teratogenic
- Does NOT inhibit cytochrome P450 enzymes responsible for synthesis of androgens
- Adverse effects:
- Nausea, vomiting, gastric pain, headache, and rashes
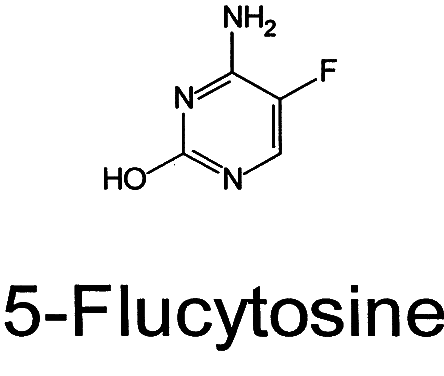
Flucytosine
Antifungal
Description
- Synthetic fluorinated pyrimidine
- Limited antifungal spectrum compared to Amp B
- Effective against:
- Candida, Cryptococcus
- Uses:
- Effective for treatment of systemic mycoses
- Meningitis caused by Cryptococcus neoformans & Candida albicans
Mechanism of Action
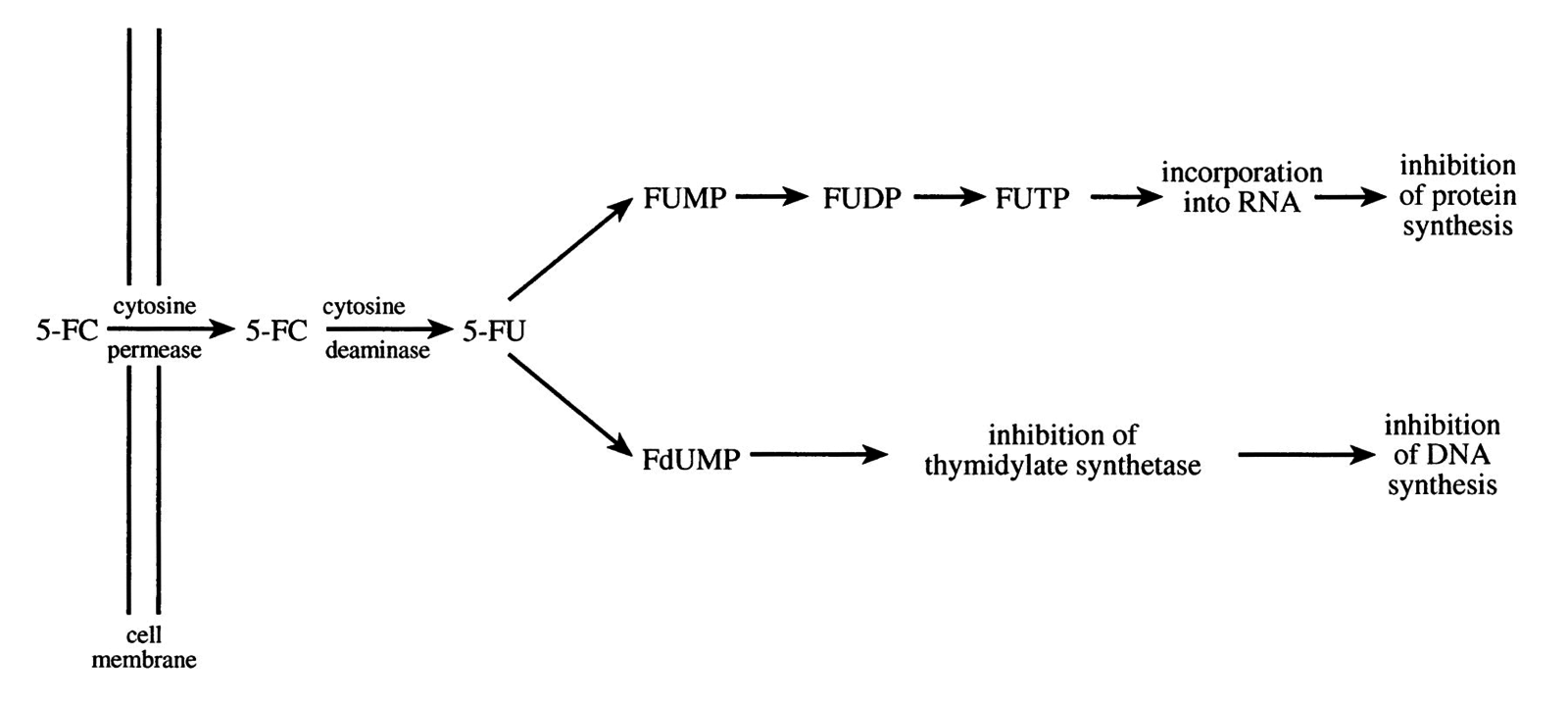
- Fluorocytosine is taken up by fungal cell via cytosine permease
- Inside the cell, it is rapidly deanimated into 5-fluorouracil (5-FU) by cytosine deaminase
- 5-FU is incorporated into DNA and then converted to 5-fluorodeoxyuridine acid monophophate
- This "false nucleotide" competitively inhibits thymidylate synthetase, thus depriving fungal cell of thymydylic acid (an essential DNA component) ultimately i nterfering w/ DNA synthesis
- I nhibits protein synthesis by being converted into 5-fluorouridine triphosphate, which is incorporated into fungal RNA, thus disturbing the amino acid pool and preventing protein synthesis
Pharmacokinetics
- Well-absorbed in the GI tract
- Eating food with flucytosine slows absorption rate
- Low affinity for serum proteins
- Peak plasma concentrations attained within 1-2 hours after oral administration
- Half-life: 3-6 hours
- Elimination occurs mostly through the kidneys
-
In patients w/ impaired renal function, higher serum levels are present and the drug accumulates
- Up to 96% is eliminated as unchanged drug
- Drug concentrations in the spleen, heart, liver, kidney, and lung are equal to those found in the blood
- 5-FC levels in the CSF are ~65 to 90% of that in the blood
Drug Interactions
- Aluminum hydroxide
- Magnesium hydroxid
- Cytosine arabinoside
- Amphotericin B
- NSAIDs
- Trimethroprim
Drug Interactions
- Aluminum and magnesium hydroxide delay absorption
- Cytosine arabinoside (cytostatic agent), has been reported to inactivate the antifungal activity of Flucytosine via competitive inhibition
- Drugs which impair glomerular filtration may prolong half-life of Flucytosine
-
Amphoterecin B is commonly prescribed with Flucytosine for therapy against cryptococcal meningoenceophalitis and severe cryptococcal pneumonia
- Amphotericin B increases cell permeability, allowing more 5-FC to enter the cell
- 5-FC and Amphotericin B are synergistic
Additional Information
-
Humans do NOT have the cytosine permease enzyme
- Selective toxicity
- In the US, flucytosine is only available as an oral capsule
-
Mechanism of resistance
- Decrease flucytosine uptake by altered permease
- Decrease synthesis of active nucleotide metabolites
-
Major toxicity
-
Depression in the bone marrow leading to
- Anemia, leukopenia, and thrombocytopenia
-
Depression in the bone marrow leading to
- Flucytosine may interfere with certain laboratory tests (including serum creatinine), possibly causing false test results
-
5-FU is suspected of being capable of producing congenital anomalies in humans
- Category C drug
- 5-FC is relatively contraindicated in pregnant women
Sedative Drugs
Steph Colaiacovo
Diazepam
Sedatives
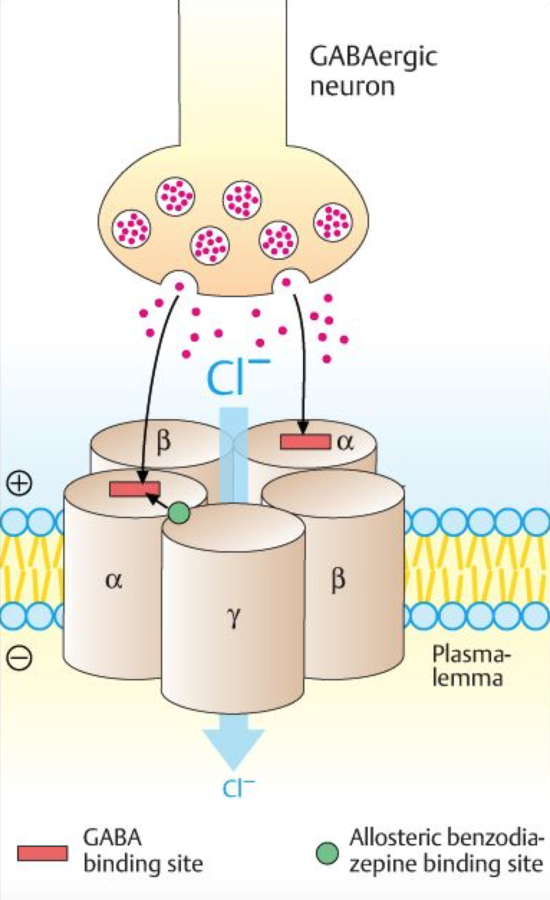
Mechanism of Action
- Potentiates GABA on the GABA receptor
- bind to benzodiazepine binding sites on GABA receptor
- Channel opens, Cl conductance increases
- neuronal inhibition results from hyperpolarization
- benzodiazepines increase the frequency that the channel opens
Uses
- anxiolytic
- muscle spasms
- seizures
- insomnia
Pharmacokinetics
- oral (Valium), IV, IM suppository
- rapidly absorbed orally
- peak plasma level: 30-90 min
- lipid soluble; crosses BBB, placenta and breastmilk
- stored in adipose tissue
- bound to plasma protein (98%)
- hepatic metabolism via CYP 3A4 and 2C19
- metabolites: desmethyldiazepam, tamazepam
- metabolites converted to oxazepam
- glucuronidation and excretion in urine
Drug Interactions
- taking sedatives simultaneously potentiates the effects of the sedatives
- alcohol: decreases metabolism increasing the sedative effects
- oral contraceptives and phenytoin: decrease elimination of metabolites; diazepam acts longer
- rifampin and carbamazepine: increase the metabolism by inducing metabolic enzymes in the gut and liver
Drugs that may interact
- sedatives (benzodiazepines, barbiturates)
- alcohol
- rifampim
- carbamazepine
- phenytoin
- sodium oxybate
- St. John's Wort
- erythromycin
- clarithromycin
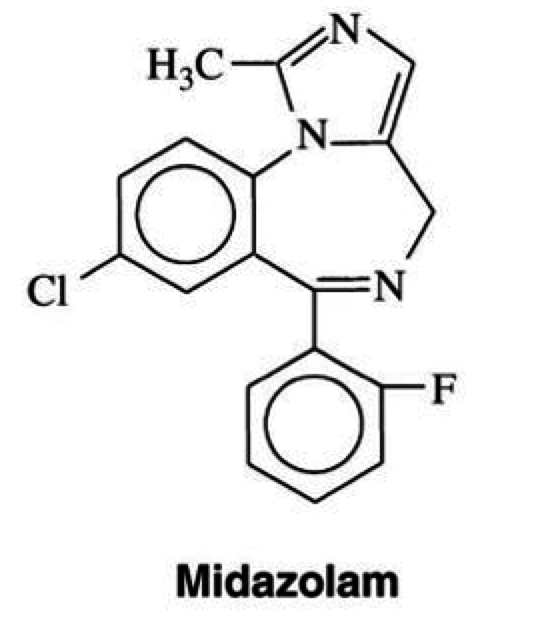
Midazolam
Sedatives
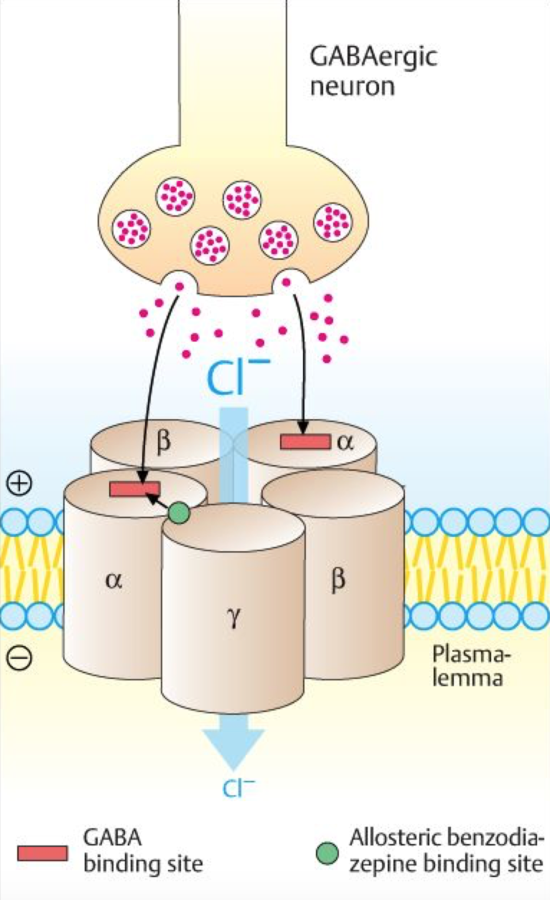
Mechanisms of Action
- potentiates GABA on the GABA receptor
- increase Cl conductance
- neuronal inhibition form hyperpolarization
Uses
- amnesic sedation
- insomnia
- anticonvulsant
- manage anxiety
- induction of general anesthesia
Pharmacokinetics
- short-acting; half life of 1-4 hours
- poorly absorbed orally; <50% reaches bloodstream
- hepatic metabolism:
- CYP 3A4
- active metabolite: alpha-1-hydroxymidazolam (less active)
- conjugation by glucuronidation
- excreted in urine
Drug Interactions
- erythromycin/clarithromycin: inhibit metabolism
- increase duration of effects
- grapefruit/ Ca Channel Blockers
- decrease CYP 3A4
- prolong sedative effect
- St. John's Wort/phenytoin/rifampin
- increase metabolism of midazolam
- less effective
- simultaneous administration of numerous sedatives potentiates effects
Drugs that may interrupt
- alcohol
- erythromycin
- aspirin
- acetaminophen
- buprenorphine
- clozapine
- tenofovir
- fluconazole
- caffeine
Phenobarbital
Sedatives
Mechanism of Action
- potentiates GABA on the GABA receptor
- allosteric binding to barbiturate binding sites
- increase conductance of Cl
- hyperpolarization, neuronal inhibition
- narrow therapeutic window
- increase the duration of channel opening
Uses
- anticonvulsant
- preferred drug for neonatal seizures
- sedative
Pharmacokinetics
- long acting; lipid insoluble
- enters BBB slowly
- onset is 1-3 hours
- duration is approx. 10 hours
- bioavailability of 90% with oral administration
- peak plasma concentration after 8-12 hours
- hepatic metabolism
- hydroxylation with cytochrome P450 enzymes
- glucuronidated and excreted in urine
- 20-50% excreted unchanged
Drug Interactions
- alcohol: together they produce severe drowsiness and psychomotor impairment
- "painless" death
- long term use of barbiturates
- increase in hepatic microsomal enzyme activity
- increase metabolism of anticoagulants (ex. warfarin)
- reduction in their function
- concomitant use of sedatives increases the sedative properties of the drug
Other drugs that may Interact
- acetaminophen
- oral contraceptives (ethinyl estradiol)
- buprenorphine
- warfarin
- lidocaine
- prilocaine
- propoxyphene
- telaprevir
Additional Information
- Contraindicated in people with porphyrias
- i.e. acute intermittent porphyria, coproporphyria and porphyria variegata
- inability to metabolize porphyrin due to a lack of an enzyme
- barbiturates increase the production of alpha-aminolevulinic acid synthase, required in the production of the porphyrin ring
- products from this synthase build up as individuals lack the enzyme that works downstream to the synthase
Chloral Hydrate
Sedatives

Mechanism of Action
- non-barbiturate sedative-hypnotic
- it enhances the inhibitory effects of the GABA receptor complex by binding to its allosteric site
- narrow therapeutic index
Uses
- liquid preparation for sedation in uncooperative children for painless technical procedures
- used in combination with nitrous oxide for sedation
- used with promethazine to relieve nausea and vomiting symptoms
Pharmacokinetics
- absorbed well with oral and rectal administrations
- metabolized in liver
- converted to trichloroethanol: responsible for CNS depressant effects
- further metabolized to dichloroacetic acid and trichloroacetic acid
- half-life approximately 4-12 hours
- glucuronidated and excreted in urine
- long term use:
- induction of liver enzyme activity
- competition for plasma protein binding sites
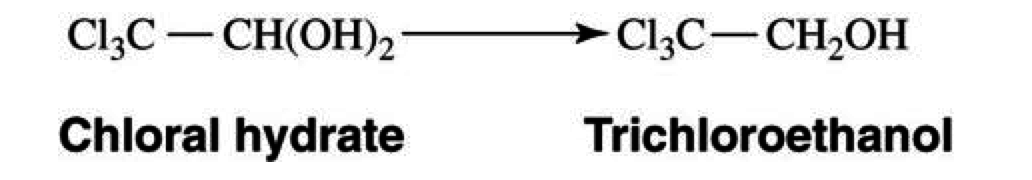
Drug Interactions
- alcohol: chloral hydrate and its metabolite inhibit the alcohol-metabolizing dehydrogenases
- blood concentration of alcohol increaases
- severe alcohol intoxication characterized by stupor, coma or death
- such intoxication referred to as "Mickey Finn" or "knock-out drops"
- trichloroacetic acid can increase the plasma concentration of warfarin
- interferes with protein binding by displacing it
Drugs that may interact
- loop diuretics (ex. furosemide)
- anticoagulants (ex. warfarin)
- H1 antagonists (ex. astemizole, terfenadine)
- albuterol
- phenylephrine
- aspirin
Narcotic Analgesics
Olga Degtyareva
Morphine
Narcotic Analgesic
Mechanism of Action
Morphine primarily acts on the mu receptor of neurons to produce analgesia. It is a G-protein coupled receptor that inhibits the action of adenylyl cyclase, which reduces the amount of available cAMP. Reduced cAMP reduces the amount of available neurotransmitter release, therefore reducing the amount of pain sensed.

Pharmacokinetics
- Peak plasma levels:
- PO - 30 min; SC - 10-30 min; IV - 5-10 min
- After first pass metabolism only 17-33% is bioavailable
- 20-35% of the drug is bound to protein in the bloodstream, and it readily crosses the BBB
- Metabolized in the liver UGTB27 into morphine 6-glucoronide and morphine 3-glucoronide
- The metabolites are then excreted primarily in the urine, but some are found in the feces as well.
Drug Interactions
- CNS depressants increase the risk of respiratory depression, hypotension, and coma
- Morphine may interact with muscle relaxants and increase their action, leading to respiratory depression
- Also drugs that inhibit or activate UGTB27 will affect the proper dosing of morphine.
- Barbituates
- Benzodiazepines
- Hypnotics
- TCA
- General anesthetics
- MAO-I
- Antihistamines
- Alcohol
- Acetaminophen
Did You Know?
- The metabolite morphine 6-glucoronide is more potent than morphine, so the dose needs to be adjusted for people with liver cirrhosis or renal failure.
- Morphine releases histamine in peripheral tissues leading to vasodilation and sensation of warmth.
- Morphine is occasionally used to treat pulmonary edema, but the mechanism for how this is achieved is unclear.
Oxycodone
Narcotic Analgesic
Mechanism of Action
Oxycodone is a semisynthetic opoid. Its mechanism of action is very similar to that of morphine (please see previous slide).

Pharmacokinetics
- Peak plasma levels: 10-15 minutes
- First pass metabolism is low, leaving 60-80% of the drug bioavailable
- 45% of drug is bound to protein
- Metabolism occurs in the liver through CYP3A4 and CYP2D6 (which results in oxymorphone, a more potent metabolite).
- The drug is then excreted in the urine, 20% of it remains unchanged, but the majority gets removed as metabolites
Drug Interactions
- Oxycodone is a CNS depressant, so caution should be taken when administering other CNS depressants because there is an increased risk of respiratory depression, hypotension, and coma.
- Oxycodone may interact with muscle relaxants and increase their action, leading to respiratory depression.
- Drugs the inhibit CYP3A4 and 2D6 may lead to increased plasma levels of oxycodone, while drugs that induce these enzymes will have the opposite effect.
- Sedatives
- Hypnotics
- General anesthetics
- Alcohol
- Diuretics
Methadone
Narcotic Analgesic
Mechanism of Action
Methadone is strictly a mu receptor agonist. Please see morphine for mechanism of action.

Pharmacokinetics
- This drug is very unpredictable – after ingesting it orally the peak plasma level may occur 4 hours later or even days later. It may last anywhere between 4 and 48 hours as doses are repeated, but it has a narrow therapeutic range so accumulation is dangerous.
- 90% of this drug is plasma protein bound, and it has a half-life of 8-59 hours.
- It gets metabolized in the liver by CYP3A4, 2B6, and 2C19.
Drug Interactions
Drugs that inhibit CYP3A4 or prolong the QT interval need to be used with caution. Increasing the accumulation of this drug or putting the patient at higher risk for arrhythmia needs to be avoided.
- Protease inhibitors
- Macrolides
- Antifungals
- Calcium channel blockers
- Diazepam
- Amiodarone
- Cyclosporine
- Haloperidol
- Antipsychotics
- Antiarrhythmics
- Antidepressants
Did You Know?
Methadone has been associated with QT prolongation and torsades de pointes.

Meperidine
Narcotic Analgesic
Mechanism of Action
- Meperidine has a very similar mechanism of action to morphine, please see previous slide.
Pharmacokinetics
- This drug is not very effective when taken orally because of a very high first pass metabolism
- It is normally prescribed as IV, and it gets metabolized in the liver to normeperidine
- The half-life of meperidine is 2.5-4 hours, but that of normeperidine is 15-30 hours, so administration to a patient with renal failure is ill-advised because the metabolites are excreted in the urine
Drug Interactions
If prescribed with MAO inhibitors, may result in serotonin syndrome. High levels of serotonin present as increased heart rate, sweating, hallucinations, and coma.
- MAO-I
- Acetaminophen
- Aspirin
- Acyclovir
- Tramadol
- Phenytoin
Did You Know?
Meperidine was once widely used for labor and delivery but has now grown out of favor
Local Anesthesia
Saj Arora
Lidocaine
Local Anesthetics
Description
- Xylocaine (lidocaine HCl) is a local anesthetic agent that is administered into the submucosa near the nerves that you want to block
- It comes in injections with or without epinephrine
- Lidocaine is usually prepared in a nonpyrogenic, sterile solution of sodium chloride
- It is crucial that the pH of these solutions is adjusted to approximately 6.5 (5.0–7.0) as uptake into neuronal tissue is highly dependent on the concentration of the uncharged molecule
- This uncharged form is more prevalent at a basic pH.
Mechanism of Action
- Lidocaine is an amide local anesthetic that blocks voltage-gated sodium channels
- These sodium channels are located in the cell membranes of neurons to generate and propagate electric current
- By i nhibiting the ionic fluxes required for the initiation and conduction of impulses, the sensation from the area can be stopped.
Pharmacokinetics
Absorption
- Local anesthetic agents, unlike most other other drugs should be absorbed poorly.
- The key to have anesthesia in one area is to keep the drug in that location.
- Therefore, a drug that has bad absorption is more effective in terms of local application.
- Lidocaine’s absorption is highly dependent on the tissue environment and the presence of a vasoconstrictor.
Pharmacokinetics
Distribution
- Lidocaine is widely distributed and it can passively cross the blood-brain barrier and the placenta
- Moreover, a large quantity (60-80%) of lidocaine is bound to proteins in the blood
- Binding also depends on the presence of alpha-1-acid glycoprotein
- The elimination half-life of lidocaine HCl following an intravenous bolus injection is typically 1.5 to 2.0 hours
Pharmacokinetics
Metabolism and Excretion
- Liver metabolism is crucial to the excretion of lidocaine
- Most of the drug is excreted by the kidney in either the metabolized (90%) or the unchanged form (10%)
- Biotransformation of lidocaine is a multi-step process that includes oxidate N-dealkylation, ring hydroxylation, conjugation and cleavage of various other bonds
- One of the issues in this metabolism is that creation of monoethylglycinexylidide metabolites which can be almost as potent as lidocaine
Drug Interactions
- Methemoglobinemia is caused by oxidation of hemoglobin resulting in decreased oxygen carrying capacity
- Symptoms include gray discoloration in the patient, light-headedness, fatigue and headache
- Sodium Nitrite is an oxidizing agent which has the capacity to oxidize hemoglobin
- Similarly, prilocaine is hydrolyzed to an o-toluidene which again is a strong oxidizer. Therefore, these interactions should be avoided.
- Possible Oxidizing Agents such as sodium nitrite, thiosulfate and other Local anesthetic agents such as prilocaine
Additional Information
-
Liver dictates the rate at which lidocaine is metabolized
-
Half-life can be two to three fold longer in patients with hepatic disease
-
Renal dysfunction can lead to build-up of metabolites
-
Bolus injections given intravenously can be very dangerous
-
CNS toxicity become very apparent with lidocaine plasma levels above 6.0μg per mL
-
Convulsions have been show in rhesus monkey at 18–21 µg/mL
-
I njections should be given only after aspiration and over a period of 2-3 minutes per carpule
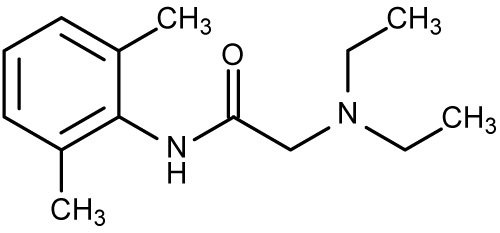
Benzocaine
Local Anesthetics
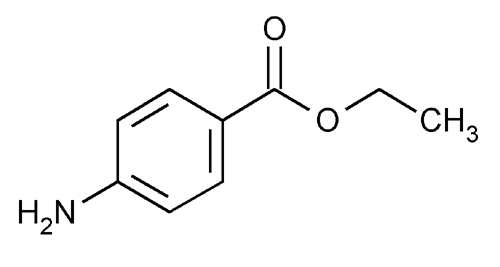
Mechanism of Action
- Benzocaine is one of the few ester anesthetic agent used today
- Applied as a topical anesthetic in water soluble base of polyethylene glycol.
- Benzocaine stabilizes the membrane and the voltage-gated sodium channels keeping them closed
- Therefore, there is no migration or generation of current in the neuron
- This in turns, blocks sensory signals downstream from reaching the brain
- The anesthesia is produced fairly rapidly and lasts about 30-60 minutes.
Pharmacokinetics
Absorption
- Benzocaine is primarily used as a topical agent
- Most of the absorption is minimal and happens through the mucous membrane
- The amount absorbed varies significantly depending on the tissue environment and location.
Distribution
- Benzocaine is similar to lidocaine in that it binds plasma proteins at a high concentration
- However, there are plasma cholinesterases that metabolize benzocaine into PABA-containing metabolites
Pharmacokinetics
Distribution (cont'd)
- These can be very toxic and this is one of the primary reasons a lot of anesthetics used today are amides.
Metabolism and Excretion
- Excretion is primarily via the renal system in the form of metabolites.
Drug Interactions
- Similar to Lidocaine, esters have drug interactions with agents that can oxidize hemoglobin
- This can lead to methemoglobinemia which may develop hours after your receive sodium nitrite or prilocaine and have been in contact with benzocaine. However, the condition can be very fatal.
-
Possible Oxidizing Agents such as sodium nitrite, thiosulfate and other Local anesthetic agents such as prilocaine
Articaine
Local Anesthetics
Mechanism of Action
- Articaine is a local anesthetic administered subcutaneously or into the submucosa so as to block nerve conduction which in turn blocks sensation
- Articaine can come preparation of 1:200,000 or 1:100,000 strength epinephrine or no epinephrine
-
Articaine works very similarly to lidocaine
-
It blocks nerve conduction and generation by blocking voltage-gated sodium channels
-
Epinephrine is a vasoconstrictor added to articaine HCl to slow absorption into the general circulation which prolongs the effects of the anesthetic, decreases absorption into blood, and decrease bleeding.
Pharmacokinetics
Absorption
- After an injection of articaine (1:200,000 epi) via the submucosal route, the peak in blood for the drug is about 25 minutes
- Whereas given three doses it takes approximate 48 minutes
- It is important to aspirate and to inject slowly so as not to give a bolus, intravenous injection.
Distribution
- Approximately 60 to 80% of articaine HCl is bound to human serum albumin and γ-globulins at 37°C in vitro .
Pharmacokinetics
Metabolism
- Articaine is first metabolized by the plasma carboxyesterase to its primary metabolite, articainic acid
- Articainic acid is inactive
- Liver uses enzymes such as P450 to further metabolize the acid
Excretion
- Half-life of articaine is 43.8 minutes with 1:100,000 epi
- Primary route for excretion of articaine is via the renal system (53-57%)
- Articainic acid is the primary metabolite in urine
Drug Interactions
- Like lidocaine, with articaine you have to worry about two types of interactions.
- First, there are drugs that will interact with the anesthetic agent (articaine).
- These include drugs that are good oxidizers like sodium nitrite and prilocaine.
- These can result in methemoglobinemia which is a dangerous condition.
Drug Interactions
- Second, there are drugs and conditions that will interact with the epinephrine packaged with the articaine.
- Epinephrine is a vasoconstrictor of the blood vessels to the smooth muscles and it is a vasodilator of the skeletal muscle vasculature.
- It also increases heart rate and the force of contraction.
- This is extremely useful for fight or flight response.
Dental Implications
- Dental use for epinephrine is for its property of a smooth muscle vasculature constriction.
- This decreases the absorption or the anesthetic and prolongs its effects.
- Tricyclic antidepressants such as Doxepin can modulate the amount of neurotransmitters that are circulating.
- Hence, epinephrine use with patients already on either uptake inhibitors or TCA should be carefully researched.
Drugs That Could Interact
- TCA’s such as doxepin (epinephrine)
- Oxidizing agents: sodium nitrite, thiosulfate
Vasoconstrictors
Albert Cheng
Epinephrine
Vasoconstrictor

Mechanism of Action
(Vasoconstrictor in general)
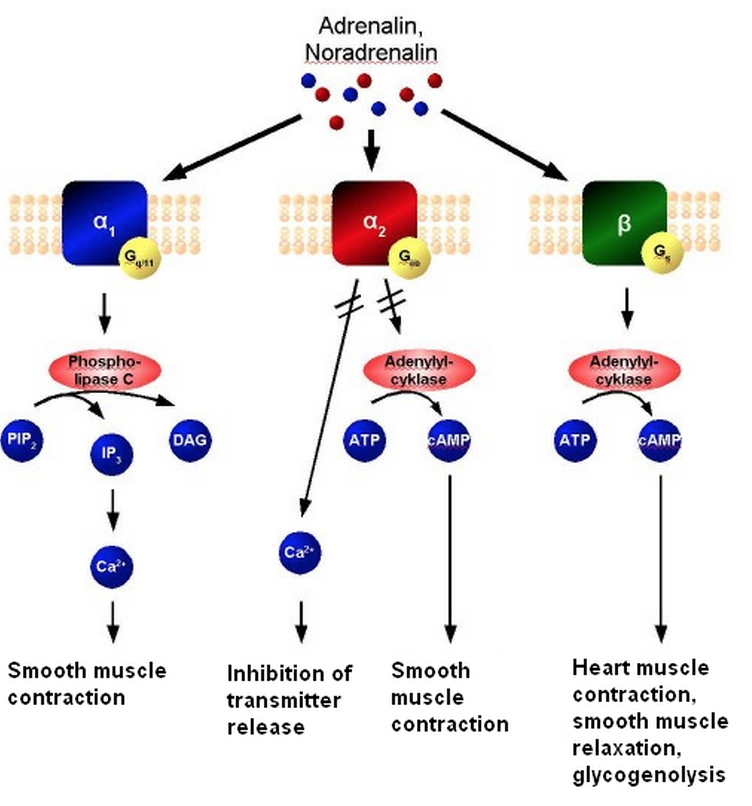
Mechanism of Action
(Vasoconstrictor in general)
-
Acts non-selectively on the alpha (@ high doses) and beta adrenergic receptors of sympathetic nervous system
-
When epinephrine binds to alpha-1 receptor, epinephrine activates G-protein subunit (Gq). The subunit
activates Phospholipase C, which cleaves membrane phospholipids into IP3 and DAG leading to an
increase in calcium
- This results in smooth muscle contraction, mydriasis, vasoconstriction in the skin, mucosa and abdominal viscera & sphincter contraction of the GIT and urinary bladder
-
When epinephrine binds to alpha-1 receptor, epinephrine activates G-protein subunit (Gq). The subunit
activates Phospholipase C, which cleaves membrane phospholipids into IP3 and DAG leading to an
increase in calcium
Mechanism of Action (cont. )
- Acts non-selectively on the alpha (@ high doses) and beta adrenergic receptors of sympathetic nervous system
-
When epinephrine binds to alpha-2 receptors, the G-protein subunit (Gi) is activated. This results in the
inactivation of adenyl cyclase and the
decrease in cAMP.
- In addition, it limits the release of neurotransmitters
-
When epinephrine binds to beta-receptors, the G-protein subunit (Gs) is activated. This results in the
activation of adenyl cyclase and
increases the level of cAMP
- This results in heart contraction, smooth muscle relaxation and glycogenlysis
Pharmacokinetics
- Metabolized in the liver by enzymes to metabephrine or normetanephrine
- Then, it is conjugated and excreted in urine
- Mostly excreted in urine as inactive metabolite and the rest as unchanged form or conjugated form
- Half-life: 2 minutes
- Onset of action (depending on route of administration)
- Subcutaneous: 5-10 mins
- Inhalation: 1-5mins
- Duration varies from 1-6 hours depending on RoA
Drug Interactions
- Alpha-adrenergic blockers (e.g. phentolamine)
- Beta-adrenergic blockers (e.g. propanolol)
- Antihistamines
- MAO/COMT inhibitors
- Tri-cyclic antidepressants
- Diuretics
- General anesthetic agents (e.g. halogenated gases)
- Levothyroxine
- Anti-arrhythmic drugs
- Digoxin
- Ergot alkaloids
Drug Interactions
- Effects may be potentiated when epinephrine interacts with MAO/COMT inhibitor, tri-cyclic anti-depressants, antihistamines, levothyroxine, and linezolid
- MAO/COMT enzymes usually degrades epinephrine after reuptake into neuronal cell
- Inhibiting MAO/COMT will prolong the effects of epinephrine on the adrenergic receptors
- Adminstration of epinephrine with halogenated gases can increase risk of cardiac arrhythmia
- Beta-adrenergic blocker can block the beta-adrenergic effects of epinephrine and lead to hypertension
- Ergot alkaloids can antagonize the vasoconstrictor effects of epinephrine
Implication in Dentistry
- One of the most popular vasoconstrictor used in anesthetic procedures for dentist
- Epinephrine helps limit the toxicity of local anesthetic agents and prolong the anesthetic effects by:
- Vasocontriction of blood vessels near the site of injection
- Limits systemic permeability
- Should be limited or avoided in patient with CVD
- Maximum dose of epinephrine is typically 500 mg (MRD) or 7.7mg/kg in an average-healthy patient
Levonordefrin
Vasoconstrictor
Mechanism of Action
- In the nasal cavity, levonordefrin binds to alpha-adrenergic receptors in the nasal mucosa and vasoconstricts to alleviate nasal congestion
- Generally, levonordefrin acts through direct alpha receptor stimulation (75%) with some beta activity (25%), but to a lesser degree than epinephrine
Pharmacokinetics
- Similar to that of epinephrine except that levonordefrin is less potent
- Elimination occurs through enzymatic degradation by MAO and COMT
Drug Interactions
- Desvenlafaxine may increase the tachycardic and vasopressor effects of levonordefrin
- We should monitor for increased sympathomimetic effects, such as
- Increased BP, chest pain, and headache when co-adminstering
Implications in Dentistry
- Levonordefrin is a safer vasoconstrictor to use in dentistry than epinephrine
- Especially for patients with CVD
- Levonordefrin is 15% as potent a vasopressor as epinephrine
- For all patient, the maximum dose is 1mg
- Primary use
- Topical nasal decongestant
- Vasoconstrictor in local anesthetic agents
Phenylephrine
Vasoconstrictor
Description
- Synthetic sympathomimetic amine
- Primary use
- Nasal decongestant
- Dilate the pupil (mydriasis)
- Increase BP
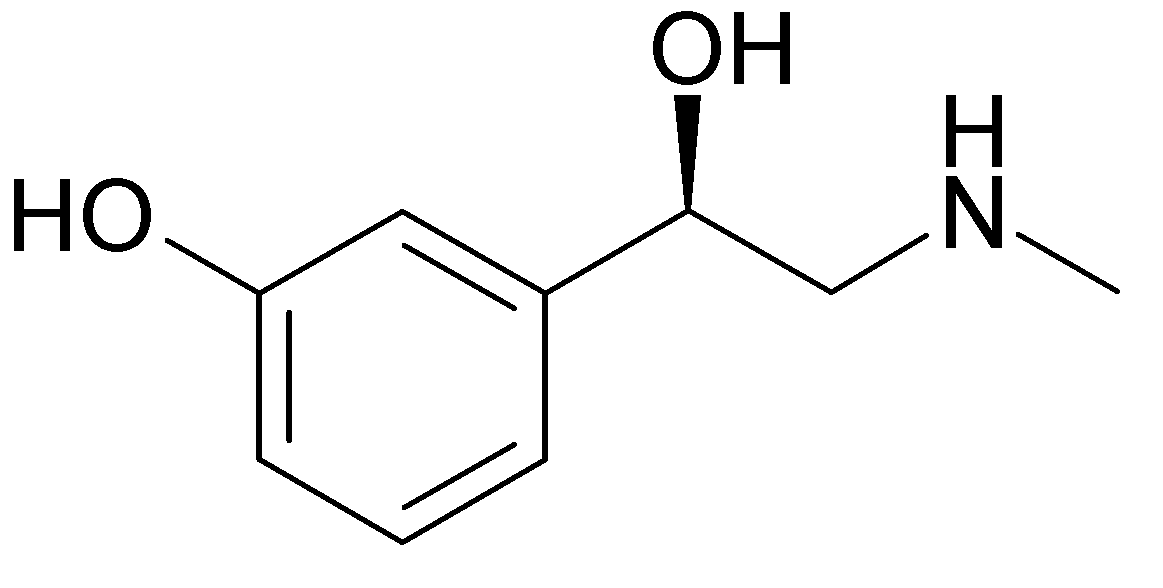

Mechanism of Action
- Relatively pure alpha-1 receptor agonist
- Avoids most of the cardiac stimulation associated with epinephrine
- May significantly elevate systolic and diastolic pressures & reflexively slow the heart for an extended period
Pharmacokinetics
- Completely absorbed through oral adminstration
- After passing through GIT, phenylephrine has 38% bioavailability
- High affinity for serum proteins and high volume of distribution into certain organ compartments
-
IV administration of phenylephrine
- Half-life: 5 mins
- Metabolized in the liver via deanimation by MAO enzymes into the major metabolite (m-hydroxymandelic acid)
- Can also undergo hydroxylation to epinephrine, then oxidation to metanephrine
- Elimination similar to epinephrine
- Only 12% of drug is excreted in its unchanged form in urine
Drug Interaction
- MAO inhibitors
- Tri-cyclic antidepressants
- Hydrocortisone
- Calcium channel blockers
- ACE inhibitors
- Benzodiazepines
- Sumatriptan
Drug Interaction
- Hypertensive effects of phenylephrine may be increased when interactions occur with MAO inhibitor, tri-cyclic anti-depressants, and hydrocortison
- Ca channel blocker, ACE inhibitors, and BZD can reduce the efficacy of phenylephrine
- Phenylephrine can worsen the side effects of migraine medications (e.g. Sumatriptan)
Implications in Dentistry
- Weakest and most stable vasoconstrictor in dentistry
- No adequate studies done on phenylephrine to determine safe and effective use in pregnant women
NSAIDs and Aspirin
Steph Colaiacovo
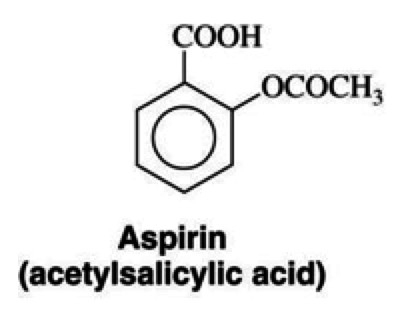
Aspirin
NSAIDs

Mechanism of Action
- aka acetylsalicylic acid
- inhibits the activity of the cyclooxyrgenase enzymes COX-1 and COX-2
- irreversibly acetylates both COX enzymes
- prevents the downstream production of the eicosanoids (ex. thromboxane, prostaglandins)
- acts on both COX-1 and COX-2, but more selective against COX-1
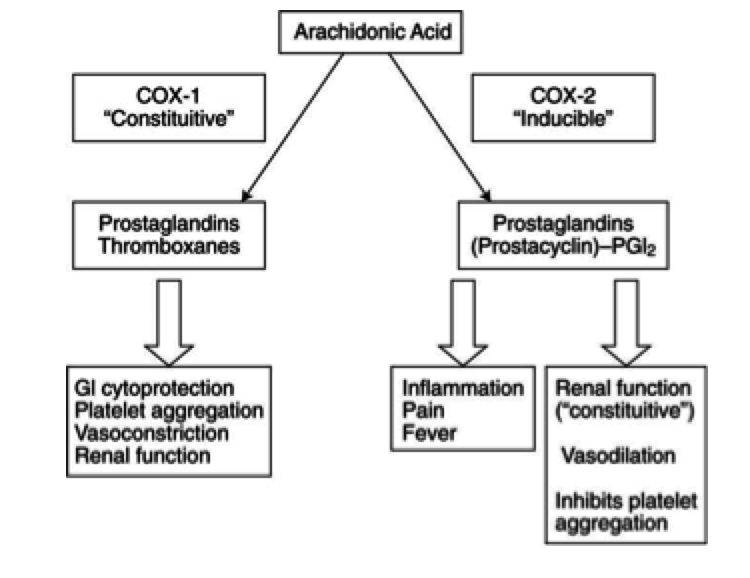
Uses
- analgesic
- antipyretic
- anti-inflammatory
- low dose (81 mg) daily:
- prophylaxis against MI and thrombotic diseases
Pharmacokinetics
- absorbed from stomach, most in small intestine
- gastric and plasma esterases convert to salicylate ion
- most salicylate is bound to plasma proteins (80-90%)
- hepatic biotransformation and conjugated into three products
- salicyluric acid
- ether or phenolic glucuronide
- ester glucuronide
- free salicylate and metabolites excreted in urine
- glomerular filtration and proximal tubular secretion
Drug Interactions
- insulin: competes with binding sites on plasma proteins
- higher among of insulin unbound in blood
- unpredictable changes in blood glucose concentration
- warfarin: aspirin displaces warfarin on plasma proteins
- internal bleeding due to platelet inactivation
- alcohol: acts on the stomach mucosa making it more sensitive to aspirin
Drugs that may interact
- ketorolac
- probenecid
- colchicine
- apixaban
- methotrexate
Additional Information
- Reye's Syndrome:
- acute onset of encephalopathy, liver dysfunction and fatty infiltration of the liver and organs
- associated with child use of aspirin
- aspirin therefore not recommended for children
Ibuprofen
NSAIDs
Mechanism of Action
- propionic acid
- nonselective inhibitor of COX-1 and COX-2
- inhibition fo eicosanoids (ex prostaglandins)
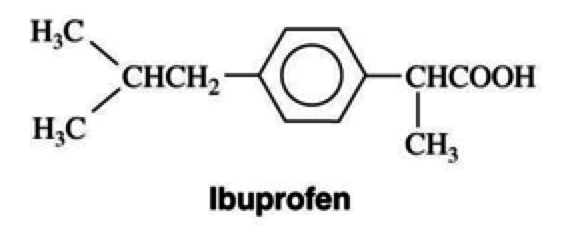
Uses
- analgesia, antipyretic, and anti-inflammatory
- preferred drug for analgesia for postoperative pain in dental extractions
- management of anti-inflammatory diseases
- ankylosing spondylitis, osteoarthritis
Pharmacokinetics
- quick onset of action, approximately 30 minutes
- highly bound to plasma albumin (99%)
- hepatic metabolism via CYP 2C9
- conjugated and eliminated in urine
- elimination half-life: 2 hours
Drug Interactions
- aspirin: decreases the anti platelet effect of aspirin
- competition for similar sites on the COX enzymes.
Drugs that may interact
- ACE-inhibitors
- diuretics
- lithium
Celecoxib
NSAIDs
Mechanism of Action
- Selective COX-2 Inhibitor
- decreases the inhibition of platelet aggregation and vasodilation effects of prostaglandins and prostacyclin
- GI protective effects form the COX-1 pathway remain but cardiovascular risks present
Uses
- analgesia
- antipyretic
- anti-inflammatory for arthritis, osteomyelitis, and rheumatoid arthritis
- familial Adenomatous Polyposis:
- reduces number of adenomatous polyps
Drug Interactions
- anti-fungals: (fluconazole and metronidazole)
- inhibits the metabolism of celecoxib by CYP 2C9
- plasma concentration of celecoxib increases
- anticoagulants: (ex. warfarin)
- severe bleeding events can occur as they both affect clotting
Other drugs that Interact
- methotrexate
- furosemide
- ACE-Inhibitors
- lithium
- aspirin
Acetaminophen
NSAIDs
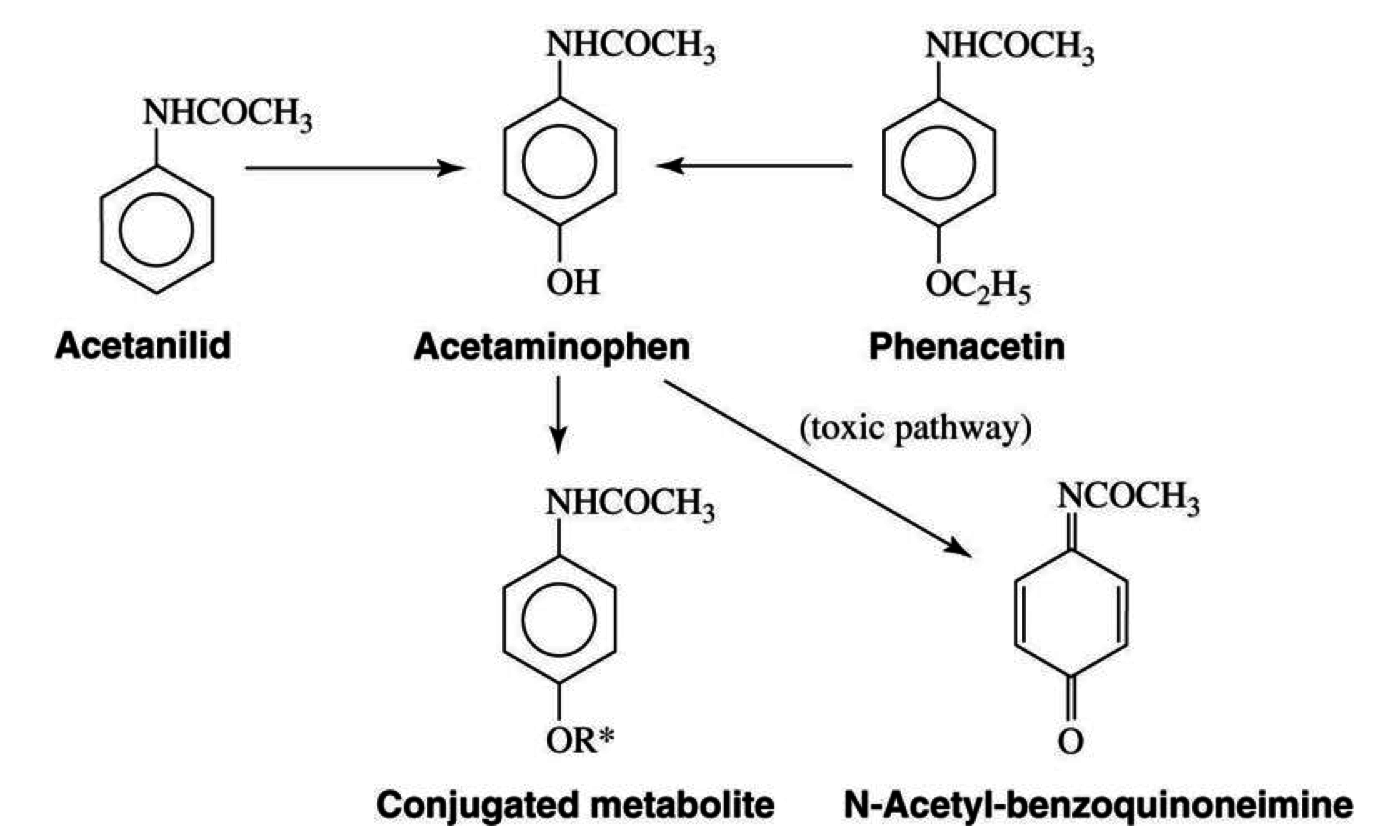
Mechanism of Action
- aka N-acetyl-p-aminophenol, tylenol
- active metabolite of phenacetin
- COX-1 and COX-2 inhibitor
- increases the threshold for pain by decreasing the synthesis of eicosanoids
- pyrogens released from leukocytes produce prostaglandins that work on the heat regulating centres in the brain
- tylenol decreases the synthesis of these prostaglandins
Uses
- treat minor aches and pains
- from colds
- headaches
- toothaches
- fever
- muscle cramps
- drug of choice when aspirin and other NSAIDs are contraindicated.
Pharmacokinetics
- well absorbed orally
- distributed throughout body fluids and tissues
- crosses the placenta
- half life: 2-4 hours
- 20-50% bound to plasma proteins
- hepatic biotransformation
- hydroxylation: 3-hydroxy-acetamin
- oxidation via CYP 2E1
- glucuronide conjuation
- conjugation with sulfate
- excreted in urine
- glomerular filtration and active proximal tubular secretion
Drug Interaction
- Alcohol: causes an induction of the CYP 2E1 enzyme
- toxic metabolite produced: NAPQI (N-acetyl-p-benzoquinoneimine)
- hepatic antioxidant glutathione is inactivated with alcohol therefore NAPQI concentration increases
Drugs that may Interact
- anti-convulsant
- diflunisal
- isoniazid
- anticoagulants
Additional Information
- Acetaminophen can cause significant hepatotoxicity after ingestion of a single dose
- single dose of 10-15g can cause hepatotoxicity
- higher doses of 20-25g can potentially be fatal
- hepatoxicity are due to the production and accumulation of NAPQI
- NAPQI causes direct damage on liver cells
- depleted liver of glutathione antioxidant
Drugs of Abuse
Dan Kim
Methamphetamine
Drugs of Abuse
Description
Methamphetamine
-
Potent CNS stimulant of the amphetamine class
-
Sometimes used to treat ADHD and obesity
-
Used recreationally to elevate mood, increase energy levels, and enhance sexual desire, allowing some users to engage in sexual activity for days
-
Schedule II drug with the following effects:
-
Euphoria
-
Reduced fatigue
-
Increased adrenergic nerve activity
-

MoA
Methamphetamine
- High lipophilicity
- Allows the drug to readily cross the blood brain barrier where it is more resistant to MAO degradation (CNS effects)
- Allows the drug to readily cross the blood brain barrier where it is more resistant to MAO degradation (CNS effects)
- Inhibition of VMAT2 receptor
- Prevents repackaging of monoamines in vesicles, resulting with elevated monoamide neurotransmitters in the synaptic cleft
- Prevents repackaging of monoamines in vesicles, resulting with elevated monoamide neurotransmitters in the synaptic cleft
- Phosphorylation of monoamine transporters
- Causes transporters to be internalized, or to work in reverse and elevate levels of monoamines in cytoplasm
- Causes transporters to be internalized, or to work in reverse and elevate levels of monoamines in cytoplasm
- Inhibition of MAO inhibitors
- Prevents monoamine degradation
-
Trace amine-associated receptor 1 (TAAR1) agonist
- TAAR1 is a GPCR that increases cAMP levels via adenylyl cyclase; this results with activation of protein kinases that phosphorylate monoamine transporters
-
Regulates catecholamine systems by non-competitive inhibition of:
- DAT - dopamine transporter
- NET - norepinephrine transporter
- SERT - serotonin transporter
Pharmacokinetics
Methamphetamine
- Directly toxic to dopamine and serotonin neurons, unlike other drugs in the amphetamine class
-
Methamphetamine PO
- Absorbed into bloodstream with peak concentrations within 3-6 hours of ingestion
-
Methamphetamine Inhaled/Intranasal
- Absorbed into bloodstream with peak concentrations at 10-24 hours
-
Metabolized by CYP2D6
-
Active Metabolites (sympathomimetics):
- Amphetamine
- 4-hydroxymethamphetamine
-
Active Metabolites (sympathomimetics):
-
Effects of gastrointestinal pH on absorption
- Acidic pH reduces absorption
- Basic pH increases absorption
-
Effects of urinary pH on excretion
- Acid pH increases excretion
- Basic pH reduces excretion
Drug Interactions
Methamphetamine
- CYP2D6 inhibitors such as SSRIs will prolong the elimination half-life of methamphetamine by inhibiting its metabolism
- MAO inhibitors increase the plasma concentration of catecholamines, resulting with a dangerous synergistic effect with methamphetamine
- Methamphetamine decreases the effects of sedatives and depressants, and increases the effects of stimulants and anti-depressants
- Methamphetamine counteracts the cardiovascular effects of anti-hypertensive drugs
- Methamphetamine counteracts the cognitive effects of anti-psychotic drugs
- Protein pump inhibitors (PPI) and H2 anti-histamines reduce gastric acid, thus increasing the absorption of methamphetamine
- Acidification of the urine with ammonium chloride is used to treat methamphetamine toxicity because it increases the rate of excretion
Drug Interactions List
- Abilify (aripiprazole)
- Adderall (amphetamine/dextraoamphetamine)
- Alcohol
- Ambien (zolpidem)
- Ativan (lorazepam)
- Celexa (citalopram)
- Cymbalta (duloxetine)
- Fish oil (omega-3 polyunsaturated fatty acids)
- Klonopin (clonazepam)
- Lyrica (pregabalin)
- Norco (acetaminophen/hydrocodone)
- Prozac (fluoxetine)
- Ritalin (methylphenidate)
- Seroquel (quetiapine)
- Tylenol (acetaminophen)
- Valium (diazepam)
- Vicodin (acetaminophen/hydrocodone)
- Wellbutrin (bupropion)
- Xanax (alprazolam)
Implications in Dentistry
- Chronic methamphetamine users show signs of mental and physical fatigue, poor oral hygiene, and facial twitching
- Of particular interest to dentistry are the worn teeth and chewed tongue that result from continuous oral movements
- Epinephrine containing local anesthetics are contraindicated in methamphetamine users because of their dangerous synergistic effect
Cannabis
Drugs of Abuse
Description
Cannabis (Marijuana)
- Most widely used illicit drug due to its high level of social acceptance and low dependence liability
-
Desired drug effects include:
- Feeling of well-being and relaxation
- Altered sensory perception
-
Adverse drug effects include:
- Psychomotor impairment
- Dysphoria, anxiety, paranoia
- Tachycardia, flushing, nausea
-
Over 60 psychoactive constituents of cannabis
- Delta-9-tetrahydrocannabinol (THC)

MoA
Delta-9-tetrahydrocannabinol (THC)
- THC binds to target receptors less selectively than "endogenous THC" aka endocannabinoids
- It is highly lipophillic allowing it to readily cross the blood brain barrier and bind non- specifically to receptors in areas of the brain, and adipose tissue
-
THC is a partial agonist of:
- CB1 receptors in the CNS
- CB2 receptors on immune cells
- Binding of cannabis to CB2 receptors is known to mediate immunosuppressant effects; but it is unclear whether cannabis produces clinically relevant immunosuprpresion
- The psychoactive effects of THC are mediated by CB1 GPCR in the CNS, which decreases cAMP through inhibition of adenylate cyclase
- Synthetic THC (marinol) is US FDA-approved for the treatment of chemotherapy-related nausea and vomiting, and appetite and weight loss associated with HIV/AIDS
- Sativex THC (buccal spray) has been developed for the treatment of neuropathic pain associated with multiple sclerosis
- Properties of therapeutic use:
- Analgesia, muscle relaxation, sedation,
- Immunosuppression, mood improvement
- Stimulation of appetite, antiemesis
- Lowering of intraocular pressure
- Bronchodilation, neuroprotection
- Induction of apoptosis in cancer cells.
Pharmacokinetics
Cannabis
- Metabolism occurs mainly in the liver by cytochrome p450 system
- CYP2C9, CYP2C19, CYP3A4
- CYP2C9, CYP2C19, CYP3A4
- Metabolites:
- 11-OH-THC has psychoactive effects and is further metabolized to 11-nor-9-carboxy-THC
- 11-OH-THC has psychoactive effects and is further metabolized to 11-nor-9-carboxy-THC
- Phase II Metabolism:
- Addition of glucuronic acid improves water solubility to facilitate excretion in urine
- Addition of glucuronic acid improves water solubility to facilitate excretion in urine
- Elimination:
- 55% excreted in feces
- 20% excreted in urine
- Elimination depends on frequency of use and body fat levels
-
Pharmacokinetics of cannabis vary as a function of its r
oute of administration
- Inhalation - fast onset of effects within 15-30 seconds; peak within 15-30 minutes; and taper off within 2-3 hours
- Ingestion - delayed onset of effects occur within 30-90 minutes; peak within 2-3 hours; and taper off within 4-12 hours
- Non-receptor dependent mechanisms allows for its variety of effects on receptors, transporters, enzymes
Drug Interactions
Cannabis
-
Smoking cannabis induces CYP1A2 metabolism
-
This increases the clearance of drugs
that are metabolized by CYP1A2
- Clozapine
- Theophylline
- Tricyclic antidepressants
- Higher doses are required when these drugs are used in combination with cannabis
- Lower doses are required if the patient seizes cannabis use
-
This increases the clearance of drugs
that are metabolized by CYP1A2
- Additive effects occur when cannabis is used in combination with
sedatives
- Significant tachycardia can occur when beta-adrenergic effects of cannabis are coupled with anticholinergic effects of
tricyclic antidepressants
- Competition occurs with other highly protein bound drugs such as
warfarin
- Cannabis-induced vasodilation of the nasal mucosa leads to increased cocaine absorption
- Cannabis enhances the onset and bioavailability of cocaine, leading to increased tachycardia
- CB1-receptor inverse agaonists (rimonabant) and opioid receptor antagonists (naloxone, naloxonazeine) can reduce the effects of cannabis
- THC is also an allosteric modulator of the mu and delta opioid receptors
- Methyllycaconitine binds to alpha-2 nicotinic receptors to stimulate cannabis reward centers without precipitating withdraw effects; this may potentially be used as a quitting aid for chronic cannabis abusers
Drug Interactions List
- Adderall (amphetamine/dextraoamphetamine)
- Alcohol
- Ambien (zolpidem)
- Ativan (larazepam)
- Cymbalta (duloxetine)
- Fish oil (omega-3 polyunsaturated fatty acids)
- Flexeril (cyclobenzaprine
- Klonopin (clonazepan)
- Lamictal (lamotrigine
- Lexapro (escitalopram)
- Lyrica (pregabalin)
- Norco (acetaminophen/ hydrocodone)
- Seroquel (quetiapine)
- Valium (diazepam)
- Vicodin (acetaminophen/hydrocodone)
- Vitamine B12 (cyanocobalamin)
- Vitamin D3 (cholecalciferol)
- Xanax (alprazolam)
- Zoloft (sertraline)
Implications in dentistry
- Cannabis use induces dry mouth (xerostomia) making users susceptible to caries
-
Chronic marijuana use may result in gingival enlargement with clinical characteristics similar to
phenytoin-induced enlargement.- (www.researchgate.net/... marijuana.../0fcfd5136659f8a3a9000000.pdf)
-
Recent inhalation of marijuana before general anethesia may cause acute uvular oedema and post-operative airway obstruction. During general anesthesia, additive effects of marijuana and potent inhaled anesthetics can result in pronounced myocardial depression, which usually accompanies severe sepsis and septic shock
-
(http://www.ncbi.nlm.nih.gov/pubmed/8807175)
-
Cocaine
Drugs of Abuse
Description
Cocaine
- Benzyolmethylecgonine (cocaine) is an alkaloid obtained from the leaves of the coca plant
- Desired effects:
- Euphoria and elevated mood
- Increasing alertness
- Feeling of supremacy
- Negative effects:
- Irritability and anxiety
- Paranoia
- Restlessness
- Signs of cocaine use
- Dilated pupils
- Excited exuberant speech

MoA
Cocaine
- Acts as a triple reuptake inhibitor (TRI) of serotonin, norepinephrine, and dopamine by blocking their transporters
-
Amplifies the natural effect of neurotransmitters on the post-synaptic neuron by inhibiting transporters that normally remove excess from the synapse
- 5-HT2 and 5-HT2 receptors
- Norepeniphrine transport protein
- D1 receptors
- Blocks NMDA receptors
- Blocks sodium channels, thereby interfering with the propagation of sodium channels
-
Binds sigma receptors to produce antidepressant-like effects
- Addiction occurs due to its effect on the mesolimbic reward pathway
Pharmacokinetics
Cocaine
- The body is not designed to function with high levels of dopamine; thus cocaine use can cause heart attack, stroke, and death
- HEART - increase in HR and BP; vasoconstriction; can trigger arrhythmias
- BRAIN - vasoconstriction of cerebral arteries can lead to stroke, seizures, and violent behavior
- LUNGS - smoking cocaine can permanently damage the lungs
- NASAL MUCOSA - snorting cocaine can cause nasal perforations
- GIT - vasoconstriction can cause oxygen starvation in the gut leading to ulcers
-
KIDNEYS - cocaine use accelerates kidney damage through rhabdomyolysis
-
LIVER -
Cocaine is rapidly metabolized in the liver, with less than one percent of the parent drug being excreted in the urine. The primary metabolite is benzoylecgonine and is detectable in the urine for up to eight days after cocaine consumption.
- FAT - cocaine directly interferes with metabolic processes that store lipids, thereby reducing body fat
Drug Interactions
Cocaine
- Deaths due to cocaine use appear to occur in a minority of those who use it; however, one study had shown an increased lethality rate when cocaine is used in combination with alcohol, heroin, opiates, antidepressant and antipsychotic medications
-
Ethanol decreases the clearance of cocaine by inhibiting the hydrolysis of cocaine to benzoylecgonine and ecgonine methyl ester by carboxylesterases
-
Co-consumption of ethanol and cocaine produces cocaethylene, which is highly toxic
- Cocaine can precipitate opioid withdraw symptoms by significantly diminishing buprenorphine concentrations, a semi-synthetic opioid used to treat opioid dependence
- Induction of CYP3A4
- Induction of P-glycoprotein
- Cocaine and acetaminophen increase serotonin levels synergistically
- Antidepressants (SSRIs) enhance cocaine-induced toxicity
- Cocaine increase the metabolism of DHEA, a drug used in the treatment of depression
Drug Interactions List
- Adderall (amphetamine/dextraoamphetamine)
- Alcohol
- Clopine (clozapine)
- DHEA (dehydroepiandrosterone)
- Geodon (ziprasidone)
- Klonopin (clonazepam)
- Norco (acetetaminophen/oxycodone)
- Oxycontin (oxycodone)
- Paxil (paroxetine)
- Percocet (acetaminophen/oxycodone)
- Prozac (fluoxetine)
- Remeron (mirtazapine)
- Ritalin (methylphenidate)
- Seroqual (quetiapine)
- Singular (montelukasf)
- Tylenol (acetaminophen)
- Valium (dizepam)
- Vicodin (acetaminophen/hydrocodone)
- Xanax (alprazolam)
Implications in Dentistry
- Cocaine has been shown to elevate plasma levels of epinephrine and norepinephrine; thus it is unsafe to administer local anesthesia with epinephrine in such patients
- Administration of gingival retraction cords impregnated with epinephrine may enhance tachycardia and elevations in blood pressure
- Cocaine can be used as a local anesthetic if applied topically
Alcohol
Drugs of Abuse

Description
Alcohol
- Alcohol is a CNS depressant that slows down vital functions, resulting with slurred speech, impaired judgment, and slowed reaction
- Alcohol has significant psychoactive effects at sublethal doses
- Medical Uses:
- Antiseptic
- Antitussive
- Antidote to methanol poisonoin
- Long-term Effects:
- Disrupt normal brain development
- Liver cirrhosis and liver disease
- GIT ulcers
- Decrease in sperm production
- Decrease iron and vitB, leading to anemia
- Alcoholism and death
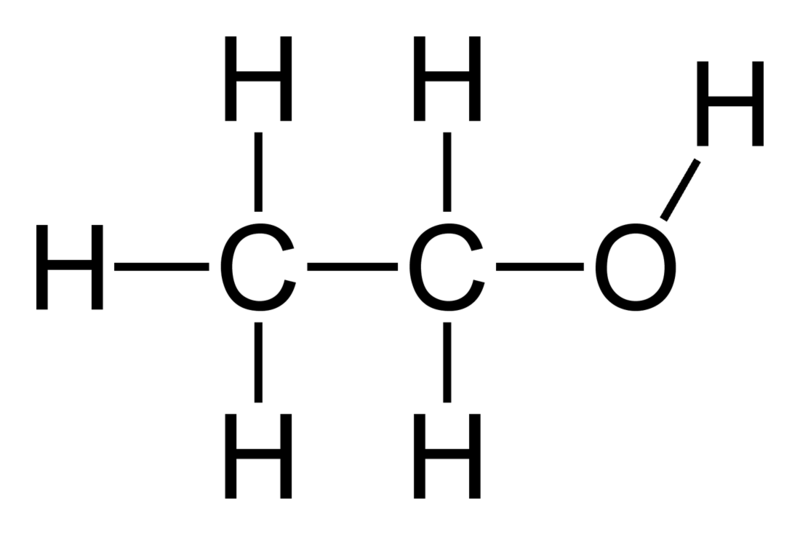

MoA
Alcohol
- Acts as an indirect GABA agonist that inhibits neuronal signaling in the CNS by increasing chloride ion influx into the post-synaptic neuron; this depresses the membrane potential and causes the sedative effects of alcohol
- Inhibits glutamate receptor to decrease excitatory neuronal signaling in the CNS
- Interestingly, chronic consumption of alcohol hyper sensitizes NMDA receptors to glutamate, and desensitizes GABA receptors
-
Negative allosteric modulator of AMPA receptor
- Crucial role in alcohol craving and relapse
-
Negative allosteric modulator of kainate reeptor
- Reduce hippocampal excitability
-
Positive and negative allosteric modulator of nAch receptor
- Important role in alcoholism
Pharmacokinetics
Alcohol
- Absorbed quickly from the digestive tract and into the blood vessels
- Alcohol is metabolized mainly in the liver by alcohol dehydrogenases (ADH) and aldehyde dehydrogenases (ALDH)
- Alcohol inhibits the brain's ability to secrete ADH, leading to decreased reabsorption of water and frequent urination
- Prolonged alcohol use induces CYP2E1
- Increases vitamin metabolism
- Increases production of reactive oxidative species
- Alcohol competes with fat for the use of NAD+, which metabolizes fat; this leads to weight gain, accumulation of body fat, and fatty liver (and cirrhosis)
Drug Interactions
Alcohol
- Exaggerates the sedation caused by other CNS depressants such as barbiturates, benzodiazepines, opioids, antipsychotics, and anti-depressants, because all of these act on the GABA receptors in the CNS
- In combination with cocaine, alcohol produces cocaethylene, a psychoactive substance with sympathomimetic properties; it increases neurotransmission in the brain by inhibiting dopamine, serotonin, and norepinephrine transporter proteins
- In combination with cannabis, alcohol increases plasma THC levels, leading to nausea, vomiting, anxiety, and paranoia
- Alcohol metabolizing enzymes (ADH and ALDH) are inhibited by disulfiram; co-consumption is used to cause acute "hangover" effects in alcoholics to deter them from drinking
- Alcohol must be avoided when taking antibacterial agents such as metronidazole, which binds ADH and ALDH, impairing the liver's ability to metabolize alcohol for excretion
- Alcohol increases risk of stomach bleeding caused by aspirin and increased risk of liver damage in patients on acetaminophen
Drug Interactions
Alcohol
- Alcohol increases the sedative effects of anti-depressants by increasing its bioavailability in the blood; exact interaction is unclear
- Alcohol has synergistic effects of drowsiness and dizziness when taken with SSRIs and MAOIs
- Alcohol is metabolized slower in women who take oral contraceptives; as a result, a women on the pill may feel intoxicated at lower doses
- Alcohol decreases the effects of beta-blockers in patient that have heart problems
- Alcohol may interact with ACEI, which is an antihypertensive used to control HTN ; however, co-consumption may drastically reduce BP and cause syncope
- Alcohol increases the risk of lactic acidosis in patients on diabetic medications such as metoformin; alcohol lowers blood-sugar levels up to 24 hours after drinking because it prolongs the effects of insulin or oral diabetic agents, leading to hypoglycemia
- Alcohol enhances the sedative effects of opioids; resulting with fatigue, drop in BP, and breathing problems
- Alcohol increases the sedative effects of sleeping aids such as Ambien
- There is no direct reaction between alcohol and PPI (protein pump inhibitors); but alcohol can erode the lining of the GIT, slow an ulcer's ability to heal, increase secretion of stomach acid, and worsen reflux in GERD patients
Implications in Dentistry
-
The risk of oral cancer is 6x higher in patients who drink alcohol compared to non-drinkers
- Epinephrine has a chronic effect in increasing alcohol dehydrogenase activity and ethanol elimination
-
Pain medications are not safe when patients have 3 drinks of alcohol daily
- Patients must stay hydrated and be monitored carefully
- Acetaminophen is hepatotoxic
- NSAIDs are nephrotoxic, but preferred
Drugs used in General Anesthesia
Brendan Ruby
Nitrous Oxide
Drugs Used in General Anesthesia
Mechanisms of Action
- Selectively inhibit NMDA receptor to decrease binding and action of glutamate
- Allows for hyperpolarization of neurons via activation of two-pore-domain K+ channels
-
Weak antagonist to the receptors AMPA, Kainate, GABAc, and 5-HT3
-
Weak agonist to GABAa and Glycine.
Pharmacokinetics
- Absorbed systemically after inhalation
- blood/gas partition coefficient of 0.47 means that it is poorly soluble in blood.
- equilibrium between the alveolar and arterial tensions is quickly reached, allowing induction and awakening to occur rapidly
- high MAC of approximately 105%
- Eliminated unchanged in the exhaled gas
- However, 0.004% undergoes reductive metabolism to nitrogen by bacteria in the gastrointestinal tract.
Drug Interactions (mechanisms of interaction)
- Use of general anesthesia and nonselective monoamine oxidase inhibitors (MAOIs) as causing hypotension or hypertension.
- Nitrous Oxide also interacts with various halogenated gases to quicken induction.
Drugs that might interact with them
- Halogenated gases
- Methotrexate
- Selegiline
- Isocarboxazid
- Phenelzine
- Tranylcypromine.
Isoflurane
Drugs Used in General Anesthesia
Mechanisms of Action
- Precise mechanism by which inhalation anesthetics produce loss of perception of sensations and unconsciousness is not known
- Meyer-Overton theory suggests that the site of action of inhalation anesthetics may be the lipid matrix of neuronal membranes
- May cause changes in membrane thickness, which in turn affect the gating properties of ion channels in neurons
Pharmacokinetics
-
Primary form of excretion is 95% excreted unchanged by exhalation
-
This compound has a low rate of biotransformation at only 0.17% of the dose being metabolized
-
Its minimum alveolar concentration (MAC) is 1.15%.
-
Blood:gas partition coefficient of 1.43
-
allows for quick induction and reversal of anesthetic
-
Drug Interactions (mechanisms of interaction)
- Increases the action of the nondepolarizing neuromuscular blocking drugs
- Potentiates the muscle relaxant effect of all muscle relaxants, most notably nondepolarizing muscle relaxants, and MAC (minimum alveolar concentration) is reduced by concomitant administration of N 2 O
- Neostigmine reverses the effect of nondepolarizing muscle relaxants in the presence of Isoflurane
- Droperidol together with isoflurane can increase the risk of an irregular heart rhythm that may be serious
Drugs that might interact with them
- Doperidol
- Neostigmine
- Nondepolarizing neuromuscular blocking agents
- Epinephrine
- Norepinephrine
- Selegiline
- Isocarboxazid
- Phenelzine
- Tranylcypromine.
Additional Information
-
Strongly relaxes vascular smooth muscle, producing a hypotension that can be useful in procedures such as surgical repair of an intracranial aneurysm
-
Pungent odor, so rarely used for induction
-
Combines the desirable cardiovascular properties of enflurane with a freedom from seizure activity and less respiratory depression and hepatic metabolism.
-
Isoflurane is chemically stable, nonflammable, and marketed in brown glass bottles.
Propofol
Drugs Used in General Anesthesia
Mechanisms of Action
- Poorly understood
- Thought to produce its sedative/anesthetic effects by the positive modulation of the inhibitory function of the neurotransmitter GABA through the ligand-gated GABA A receptors
- Considered to be a NMDA antagonist
Pharmacokinetics
- Rapid onset of action
- Half-life of 1 to 8 minutes, which results in an extremely short duration of action
- Terminal elimination half-life reported to be as short as 2 hours
- Extensively conjugated in the liver to inactive glucuronide and sulfate metabolites, with less than 0.3% of an administered dose appearing in the urine as unchanged drug
- Extensive plasma (98%) and tissue protein binding contributes partly to an enormous steady-state volume of distribution of 2 L/kg to 12 L/kg
Drug Interactions (mechanisms of interaction)
- Can significantly increase the risk of serious side effects such as respiratory depression, low blood pressure, fainting, coma, and even death
- You should avoid the use of alcohol with this drug
Drugs that might interact with them
- Selegiline
- Isocarboxazid
- Phenelzine
- Tranylcypromine
- Sodium Oxybate
Additional Information
- Most commonly used IV anesthetic
- Formulated in a lipid emulsion that contains soy and egg and has a rapid onset and short duration
- Decreases intracranial pressure which makes it desirable for neurosurgery
- Little to no post-operative nausea and vomiting and is generally well tolerated by patients.
- Disadvantages include inducing hypotension, myocardial depressant, decreased peripheral vascular resistance, apnea, allergies, and could lead to infusion syndrome
Ketamine
Drugs Used in General Anesthesia
Mechanisms of Action
- Antagonist of the N-methyl-D-aspartate (NMDA) class of glutamate receptors
- NMDA inhibition produces catalepsy, consistent with the effect of ketamine administration.
- Produces profound analgesia, which seems to be at least partially mediated by µ opioid receptors, in addition to its binding to the phencyclidine binding site on the NMDA receptor.
Pharmacokinetics
- Administered by the intravenous, intramuscular, oral, and rectal routes
-
Onset of action and peak plasma concentrations occur:
- within 1 minute after intravenous administration
- 5 to 15 minutes after intramuscular injection
- 30 minutes after oral ingestion.
- Distributional half-life ranges from 11 to 16 minutes
- Elimination half-life is 2 to 3 hours
- Highly lipid-soluble, and little binds to plasma proteins (12%)
- Orally, it undergoes first-pass metabolism , where it is biotransformed in the liver by CYP3A4 (major), CYP2B6 (minor), and CYP2C9 (minor)
Drug Interactions (mechanisms of interaction)
- Prolonged recovery time may occur if barbiturates and/or narcotics are used
- Use of many drugs with ketamine may increase side effects such as dizziness, drowsiness, confusion, difficulty concentrating, and other nervous system or mental effects
Drugs that might interact with them
- Acetaminophen
- Selegiline
- Isocarboxazid
- Phenelzine
- Tranylcypromine
- Sodium Oxybate
- Levomethadyl acetate
- Propoxyphene.
Additional Information
- Relative of the psychedelic drug phencyclidine (PCP, angel dust)
- Produces a unique state known as dissociative anesthesia
- Can cause sympathetic stimulation (increased peripheral and pulmonary vascular resistance increased heart rate)
- Can produce hallucinations (pre-medicate with benzodiazepine)
Antihistamines
Alex Pisapia
Diphenhydramine
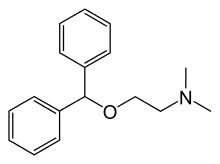
Mechanism of Action
-
Diphenhydramine is a first generation competitive H1 histamine receptor antagonist
-
Bind to the H1 receptor and reverses the effects of histamine
-
Pharmacological effects include:
-
Inhibition of the contraction of GI and bronchial smooth muscle
-
Inhibition of increased capillary permeability
-
Inhibition of flare and itch
-
Antagonize increased secretions of salivary and lacrimal glands
-
Increase release of epinephrine from the adrenal glands
-
Mechanism of Action
-
Other clinically useful effects of antihistamines that are able to cross the blood brain barrier and exhibit CNS effects include:
-
Sedation
-
Inhibition of nausea and vomiting (especially associated with motion sickness)
-
-
The mechanism of action for the CNS effects of antihistamines is not fully understood
Pharmacokinectics
- Orally administered
- Bioavailability of 40-60%
- Half life is approximately 4 hours
-
Primarily metabolized by the liver
- Hepatic injury or damage can result in altered metabolism that may vary from the norm
Drug Interactions
-
Adverse effects from first generation H1 receptor antagonists like Diphenhydramine include:
-
Drowsiness
-
Diminished alertness
-
Lethargy
-
Decreased motor coordination
-
GI disturbances such as nausea and vomiting
-
-
However, these adverse effects are relatively rare and rarely have severe drug interactions
Drug Interactions
-
Propoxyphene
-
Propoxyphene is an inhibitor of CYP450 2D6 enzyme in the liver
-
This could result in an altered metabolism of Diphenhydramine
-
-
Aspirin and caffeine:
-
Can result in altered metabolism of Diphenhydramine as well
-
Mechanism of action is the same as above
-
-
Lidocaine/Potassium Chloride
-
The mechanism of action of this interaction is the decreased gastric and intestinal motility due to the anticholinergic effects of Diphenhydramine
-
This increases potassium chlorides contact time with the gastric mucosa which can cause the adverse effects
-
Additional Information
-
General therapeutic uses for first generation H1 receptor antagonists include:
- Nasal allergies (in non-allergic rhinitis H1 antagonists are not as effective)
- Allergic dermatoses (uticaria or hives)
- The common cold (by drying the mucosa)
- Motion sickness (by inhibiting nausea and vomiting in CNS)
- Sedation, and reduction of tremors and muscle rigidity (suggestive of competitive antagonist of muscarinic Ach receptors)
- Some local anesthetic activity (suggestive of some degree of sodium channel blocking activity)
- Reduction of post operative nausea and vomiting
- Premedication for general anesthesia or deep sedation
Additional Information
- It is also important to keep in mind that H1 receptor antagonists have little effect on H2 receptors, which are mainly responsible for gastric acid secretion in the stomach
-
During anaphylactic reaction antihistamines are not the primary drug of choice because they cannot control marked hypotension and or bronchospasm associated with severe anaphylactic reaction
- For this reason epinephrine is the primary drug of choice for anaphylactic reaction
-
Antihistamines have little effect on manifestation of acute bronchial asthma
- This is due to the fact that there are other mediators involved with the bronchoconstriction seen in asthma besides histamine
- For this reason beta adrenergic receptor agonists and corticosteroids are the drugs of choice used to treat asthma
Loratadine
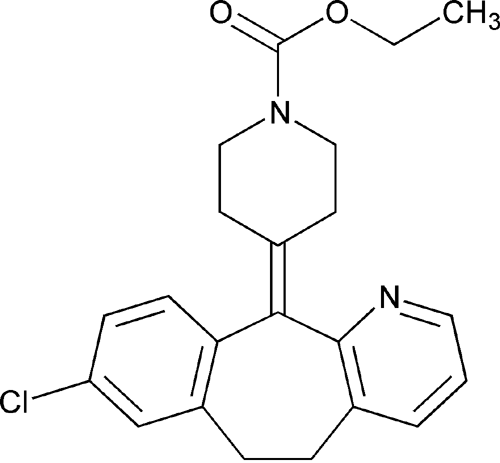
Mechanism of Action
-
Loratadine is also a competitive H1 receptor antagonist
-
However, unlike Diphenhydramine it is a second generation H1 antagonist
-
The main difference is that it does not have the sedation effect that the first generation H1 antagonists have
-
Most of the pharmacological effects of Diphenhydramine are the same as Loraradine except for the sedation effects seen in Diphenhydramine and other first generation antihistamines
-
Pharmacokinetics
-
Well absorbed in the gastrointestinal tract and has a high first pass metabolism
-
Mainly metabolized by the liver, specifically by enzymes CYP3A4 and CYP2D6
-
Approximately 97% bound to plasma proteins
-
Has a metabolite that it is broken down to called Desloratadine, which also has antihistamine effects, and is highly bound to plasma proteins
-
Half life for Loratidine is approximately 8 hours and the half life for Desloratidine is approximately 27 hours
-
Both Loratidine and Desloratidine are excreted in the urine and feces
Drug Interactions
- Most of the drug interactions with Loratidine are moderate interactions
- Amiodarone has a moderate interaction when administered with Loratidine
-
Amiodarone:
- Prolonged QT interval and torsade de pointes has been observed when these two drugs are administered together
- This is normally a side effect of Amiodarone and not of Loratidine
- The exact mechanism of action is unknown, but it is theorized that Loratidine competes with Amiodarone for the CYP3A4 enzyme in the liver which metabolizes both drugs
Additional Information
-
Loratidine causes less sedative effects when compared with other first generation H1 antihistamines
-
It crosses the blood brain barrier to a lesser extent
-
Therefore it is less likely to exert any of its effects on the CNS
-
Cimetidine
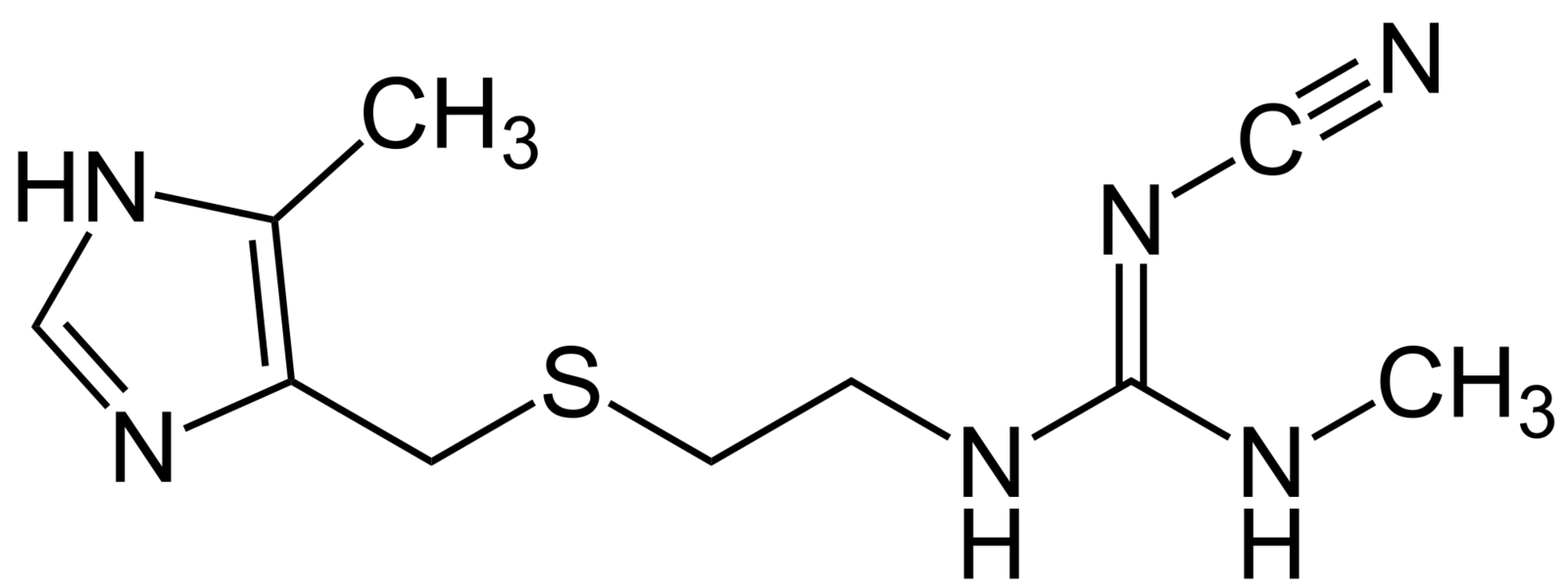
Mechanism of Action
-
Cimetidine is a competitive antagonist of the H2 histamine receptor
-
The H2 receptor is largely associated with the secretory function of the gastric mucosa
-
Antagonism of the H2 receptor results in:
-
Reduction of H+ ion output
-
Reduction in pepsin activity
-
Reduction of the total volume of gastric secretion
-
Pharmacokinetics
-
The drug can be administered orally or parenterally
-
Oral administration and parenteral administration both result in favorable therapeutic doses
-
-
The half life of Cimetidine is approximately 4-5 hours
-
The drug can be metabolized to a sulfoxide metabolite and it is mostly excreted through the urine
-
More of the parent compound, Cimetidine is recovered in the urine when administered IV
-
Drug Interactions
-
Warfarin
-
When administered together with Cimetidine increased bleeding may result
-
This adverse drug interaction is believed to occur due to Cimetidine inhibiting metabolism of Warfarin
-
Results in increased anti-coagulative effects and ultimately leads to increased bleeding
-
-
Ibuprofen/Oxycodone
-
Cimetidine’s minorly inhibits the liver enzyme CYP2D6
-
Results in decreased metabolism of Ibuprofen and Oxycodone which results in increased plasma concentrations
-
Additional Information
-
The most common therapeutic usage of H2 receptor antagonists such as Cimetidine is ability to inhibit both basal and stimulated secretion of gastric acid
-
This allows them to be used to accelerate the healing of duodenal and gastric ulcers
-
Some adverse effects of Cimetidine include:
-
Dizziness
-
Lethargy and fatigue
-
Hallucinations
-
Seizures
-
-
Although all the H2 histamine receptor antagonists have similar therapeutic effectivness, Cimetidine has the most side effects when compared with the other H2 antagonists
-
Even still, adverse effects are minimal and rare
-
Ranitidine

Mechanism of Action
-
Like Cimetidine, Ranitidine is a competitive H2 histamine receptor antagonist
-
It is hydrophilic and is unable to cross the blood brain barrier
-
Results in weak CNS and local anesthetic effects
-
-
Ranitidine also results in:
-
Decreased H+ ion output
-
Reduction in pepsin activity
-
Reduction of the total volume of gastric secretion
-
Pharmacokinetics
- Ranitidine has 50% oral absorption
- Relatively low plasma protein binding of 15%
- Ranitidine is metabolized to an N-oxide, which is an active metabolite
- Half life of approximately 2.5-3 hours
- Drug is mostly excreted in the urine
Drug Interactions
-
Atazanavir/Cobicistat
- When these protease inhibitors, which are used to treat HIV are administered with Ranitidine, reduced viral susceptibility and resistance may develop
- Because H2 antagonists such as Ranitidine decrease gastric acid secretion, this can affect the oral bioavailability and absorption of Atazanavir and Cobicistat
- When the pH of the stomach increases due to less acid secretion, the solubility of Atazanavir decreases resulting in less bioavailability
Additional Information
-
Because Ranitidine is a competitive antagonist of the H2 receptor it can be used to treat:
- Gastric and duodenal ulcers
- GERD or esophageal reflux
- Zollinger-Ellison syndrome in which too much stomach acid is produced
Food
Olga Degtyareva
Grapefruit Juice
Food
Mechanism of Action
-
Grapefruit juice irreversibly inhibits and degrades the CYP-3A4 enzyme in the small intestine as well as translation of mRNA into protein.
- It may also activate P-glycoprotein, which is a protein found in the cell membrane of enterocytes that pumps drugs out of the cell and into the lumen, which decreases the bioavailability of the drug.

Pharmacokinetics
-
The most likely constituents responsible for the interaction of grapefruit juice and other drugs are: glycosides, furanocoumarins, and sesquiterpen.
- One study showed that a single 200mL glass of grapefruit juice a day can inhibit CYP-3A4 enough to cause as much as a 9-fold increase in the peak of certain drugs. However, results vary among individuals, which makes it very difficult to predict an exact dose.
Drug Interactions
-
As a result of the inhibition of CYP-3A4, the first-pass metabolism of many drugs is decreased, and the bioavailability of these drugs is increased. Therefore, any drugs that are ingested orally and metabolized by CYP-3A4 should be avoided with grapefruit juice, especially those with active metabolites.
- There should be at least 2-3 days in between grapefruit juice intake and drugs that may be affected to prevent an interaction.
- Anti-hypertensives and anti-arrhythmics
- Antimicrobials
- Benzodiazepines
- Antihistamines and serotonin analogs
- Statins
- Chemotherapeutics
Did You Know?
- Grapefruit juice may potentiate weight loss and promote cholesterol reduction
- Some studies have shown that excessive intake of grapefruit juice may increase the risk for breast cancer in postmenopausal women.

Coffee (caffeine)
Food
Mechanism of Action
-
Caffeine is an adenosine receptor antagonist. It promotes the release of neurotransmitters and stimulates the medullary vagal, respiratory, and vasomotor centers. .
-
The overall result is an increased state of alertness, decreased drowsiness, elevated heart rate, and bronchodilation.

Pharmacokinetics
- It takes approximately 45 minutes for caffeine is absorbed by the small intestine.
- Peak plasma levels: 1-2 hours
- There is minimal first-pass metabolism.
- Average half-life is considered to be 5-6 hours
- It is metabolized in the liver by CYP1A2 into paraxanthine, theobromine, and theophylline, and eventually excreted through the kidney
Drug Interactions
Drugs that inhibit or are metabolized by CYP1A2 may lead to an increase in plasma levels of caffeine, leading to potentially toxic effects such as delirium or seizure.
- Fluoroquinolones
- SSRI antidepressants
- Antiarrhythmics
- Antipsychotics
Did You Know?
-
95% of caffeine is metabolized by CYP1A2, which makes it a great probe for the assessment of this enzyme’s activity.
- Coffee stimulates gastrin secretion and thus increases gastric acid secretion which may exacerbate symptoms in patients with peptic ulcer disease.

Licorice (glycyrrhizic Acid)
Food

Mechanism of Action
-
It inhibits 11β-hydroxysteroid dehydrogenase, which is responsible for converting cortisone to cortisol, which makes it responsible for the levels of active glucocorticoids in the body
- Licorice also inhibits short-chain dehydrogenase reductases, which stops the breakdown of stomach-protectant molecules and throwing off the balance of sodium and potassium.
Pharmacokinetics
- After first pass metabolism, glycyrrhizic acid gets converted to glycyrrhetic acid, which is what gets absorbed.
- In the bloodstream the vast majority of the compound is bound to plasma protein.
- After absorption, it gets metabolized by the liver and commensal bacteria in the gut convert it back to glycyrrhetic acid (enterohepatic cycling).
Drug Interactions
- Licorice may increase the metabolism of warfarin, thus decreasing its effectiveness, and increasing the chance of blood clots.
- Licorice may also decrease the effectiveness of hormones (like oral contraceptives) because of its effect on glucocorticoids.
- Due to the decreased levels of potassium, diuretics should be taken with caution.
- Diuretics
- Oral contraceptives
- Digoxin
- Warfarin
- Antihypertensives
- Corticosteroids
Did You Know?
- Licorice root has been used to treat GI problems, such as ulcers, heartburn, and inflammation. It has also been used for systemic lupus erythematosus, malaria, tuberculosis, chronic fatigue, and eczema.
Chocolate (tyramine)
Food
Mechanism of Action
Tyramine acts as a neurotransmitter and gets taken up into nerve terminals and causes the release of catecholamines – dopamine, norepinephrine, and epinephrine.

Pharmacokinetics
- Tyramine is a derivative of tyrosine, is found naturally in the body. It gets metabolized by monoamine oxidase
Drug Interactions
-
There is a very dangerous interaction with MAO-Inhibitors because they block the function of the enzyme. The consumption of tyramine-rich foods (like chocolate) and MAO-Is can lead to hypertensive crisis because excess levels of tyramine displace other neurotransmitters.
- Chocolate also contains caffeine, and should therefore be avoided with similar drugs.
- MAO-Is
- Ritalin
- Ambien
- ACE inhibitors
Did You Know?
-
Chocolate also contains tryptophan, which is essential for the catabolism of serotonin, and may be responsible for the feeling of overall happiness when consuming chocolate.
- High levels of tyramine are also found in certain aged cheeses, soy sauce, cured meats, yeast extract products (like beer), and fermented cabbage.

Google Doc
We tried to make this fun and we had to summarize the main points for each drug. To see our complete research please click the following link:
https://docs.google.com/document/d/194xWanHPfh6lGIeXuJCu0mQhehJn5AL0f7wvAGe0-Ms/edit?usp=sharing
this
is the
End
Thank
You
Resources
- http://www.drugs.com
- http://www.rxlist.com
- http://www.rxmed.com/
- http://www.ncbi.nlm.nih.gov/pubmed
- http://flipper.diff.org/
- http://informahealthcare.com/
**These are the most used. The rest are in the document.
Pharm Project
By Saj Arora


