Functional Analysis
of ChIP-Region
Bioinformatics Core Team
Overview
- Enrichment for gene sets
- Motif identification
Fisher or Hypergeometric testing
Often used in expression analysis, binomial, hypergeometric and Fisher enrichment tests can be used for ChIP-seq too.
- First annotate peaks to genes
- Compare peaks in genes in gene set to expected number of peaks in genes in gene set.
- The complexity is annotating peaks to the right gene and hence gene set.
Simple approach
- Select a region around TSS and promoter of consistent size.
- Promoter/TSS sizes are forced to be the same size.
- This means no gene has a unfair advantage over other genes to associated to a peak
- Longer genes
- Genes in isolation if associated by nearest method.
Simple approach
- A peak has been associated to one or more promoters/TSS it overlaps.
- The gene set test is then performed by binomial/fisher/hypergeometric test as for expression arrays.
- This can be done post Peak to Gene association in programs like David
Less simple approach
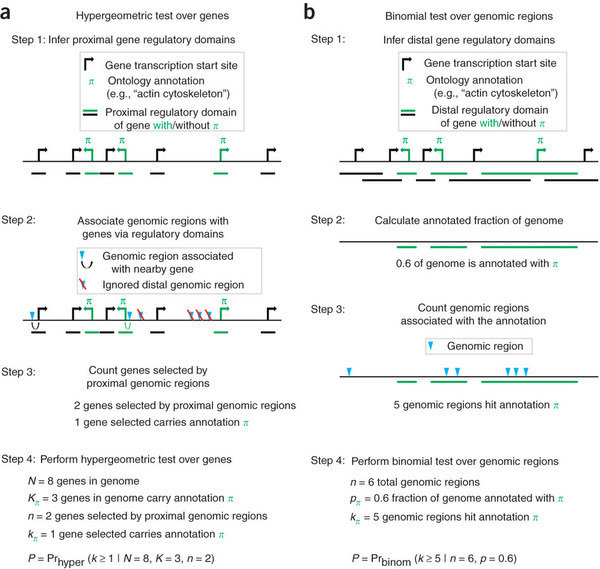
- The GREAT piece of software annotates peaks to genes regulatory regions.
- Regulatory regions are created by extending genes until they meet the regulatory region of another gene or a cut-off for distance.
- This means every gene has different chance of having an overlap with a peak.
- The GREAT software then uses a binomial test to compare peaks in gene sets regulatory regions based on the gene sets proportion of the genome.
Other tools
- There are lots of tools for ChIP-seq gene ontology analysis.
- These are just some of the most popular
- Some account for Mappability or use permutation for statistics.
Motif Enrichment
- Motif enrichment analysis can easily be split into two parts
- Denovo motif identification.
- Enrichment for known motifs
- Both require a background which is appropriate
Choosing a background
- For expression studies this is quite straight forward
- All promoters for instance
- For ChIP-seq this isn't so obvious.
- A background can be gained by scanning the whole genome.
- Or from permuting the supplied sequences to maintain sequence structure.
Choosing a database of Motifs
- Up until recently Transfac pro was the only option.
- But more recently Jaspar database got an update.
- Jaspar is freely available and is maintained as an R package by Ge Tan in Boris's group.
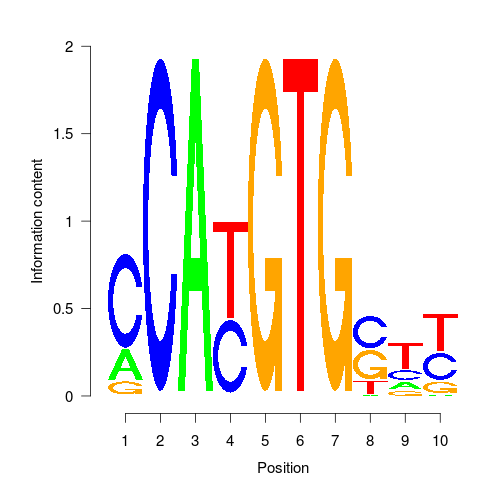
PWMS
- PWMs are point weight matrices.
- They represent the probability of a base occurring at that position for a motif.
- They are the result from Denovo motif searching and input to known motif searching
| | 1| 2| 3| 4| 5| 6| 7| 8| 9| 10| |:--|---------:|--:|--:|---------:|--:|--:|--:|---------:|---------:|---------:| |A | 0.2213683| 0| 1| 0.0000000| 0| 0| 0| 0.0091846| 0.1589503| 0.0153702| |C | 0.6841612| 1| 0| 0.4301781| 0| 0| 0| 0.4305530| 0.2037488| 0.3518276| |G | 0.0944705| 0| 0| 0.0000000| 1| 0| 1| 0.3900656| 0.1042174| 0.1501406| |T | 0.0000000| 0| 0| 0.5698219| 0| 1| 0| 0.1701968| 0.5330834| 0.4826617|
Denovo Motif Searches
- Denovo searches for denovo motifs and will find motifs most likely to be enriched in your sequence with no prior knowledge.
- Motifs discovered will be compared to a database to see if they are known.
- Often a background of scrambled sequences or Markov models from input are used.
- NestedMica and Meme-ChIP are two popular tools.
Known Motif Searches
- Search sequences for known motifs.
- All Motifs from Jaspar
- Will score success sequences against maximum score for PWM and present as proportion.
- When compared between two groups or a suitable background this can be useful.
Time for the last bit of code!
Functional Analysis
By tom carroll



