HLI WGS HG38
Summary
- VCF File Characteristics
- Sample characteristics
- High Pass Regions
- Manifest of IDs
VCF File Characteristics
- N = 2357 files
- N variants (Min,Mean,Max)
- (4,495,629; 4,738,841; 5,846,745)
- Not phased
- Not QCed
Sample characteristics
- N=2357
- Sample volume
- DNA concentration
- DNA mass
- DNA purity 260/280 ratio
| Characteristic | 1-missingness | Mean (SD) |
|---|---|---|
| Sample Volume (uL) | 100% | 60.15 (3.80) |
| DNA mass (ng) - calculated | 99.6% | 13693.10 (10186.93) |
| DNA concentration (ng/uL) | 99.6% | 228.58 (170.61) |
| DNA purity (260/280) | 17.5% | 1.72 (0.10) |
Notes:
- There is negligible protein contamination. (100% DNA -> 260/280 is approximately 1.8)
- Min mass >2220 ng, Min volume > 30 uL, Min concentration >27ng/uL
- Ilumina usually requires a minimum of 10 nM in 20 uL
- Min nM>80 assuming insert size of 50bp + 2x 151 bp paired-end nmer
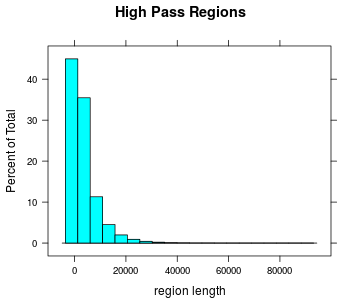
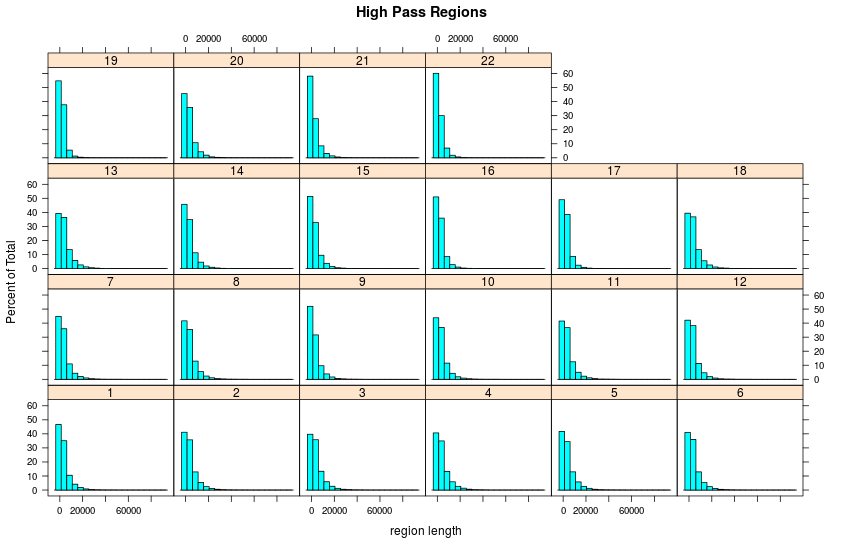
Highpass regions
- Number
- Length
- Total length
Number of regions | chr
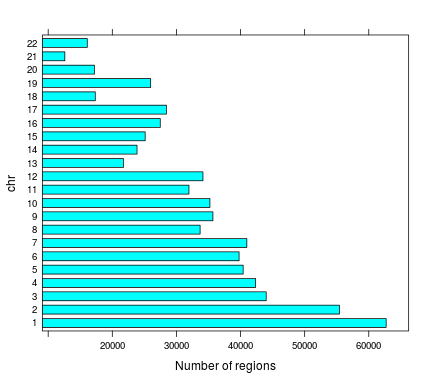
Total number of regions 712,265
Length of highpass regions
Length | chr
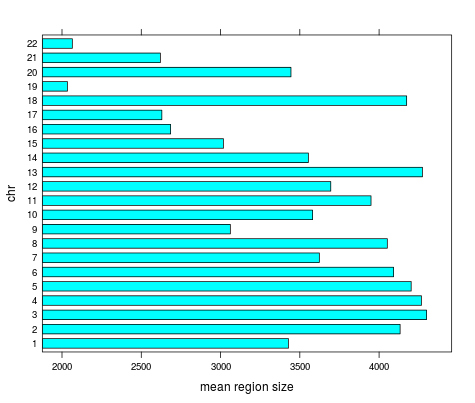
Mean length | chr
Highpass total length | chr
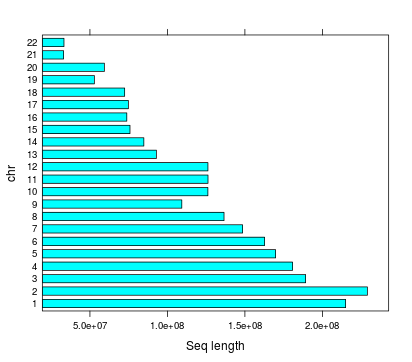
2,572,251,922 (84%)
HG38 Total non-N lenght = 3,074,968,030
Manifest of IDs
- Number of individuals sent
- Number of individuals sequenced
- Number of twin pairs and zygosity
- Number of trios
Analysis of IDs
| Samples | Retrieved | Submitted | Intersection |
|---|---|---|---|
| N | 2,357 | 2,069 | 2,036 |
| Setdiff | 321 | 33 | 0 |
| Gender | 1971/65 | ||
| Singletons | 134 | 134 (46/88) | |
| Twins pairs | 951 | 951 (401/550) | |
| Triplets | 0 | -- | |
| Trios | 159 (with DNA) | 161 (41/120) | |
| Parents | 175 (with DNA) | ||
| Missing from DB | 106 | ||
| Missing Annot | 40 |
Total = Singletons + 2xTwins + Parents = 2211
Omics overlap
| Omics | N |
|---|---|
| PainExomes | 272 |
| GOT2DExomes | 100 |
| UK10K | 861 |
| EB_Fat | 545 |
| EB_Skin | 516 |
| EB_LCL | 586 |
| EB_WB | 298 |
| Fat_450K | 449 |
Pipeline for DNASeq
Annotating, merging, coverting, validating, and quality
Filter Chr20: vcftools
Merge VCF: BCFtools
WG HLI HG38 VCF
Convert to plink: BCFtools
flip +ve strand: plink
Annotate VCF: GATK
Filter highpass regions: Bedtools
gen file
matrixeqtl file
phasing: shapeit2
GenotypeConcordance: VCFtools
Exon HG19 vcf
Coordinate change HG38:
crossmap
convert files: qctool / gtools
plink file
phased plink file
QC individuals
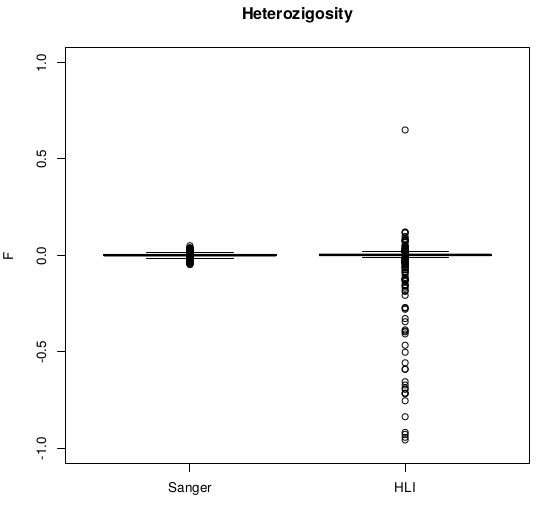
- Analysis conducted on SNPs shared by HLI WGS and Genotyping arrays merged at Sanger
- Comparison of statistics with and without exclusion of individuals with high heterozygosity (Het>0.4)
Releases comparison Hetererozygosity
Sanger vs Alex
Heterozigosity
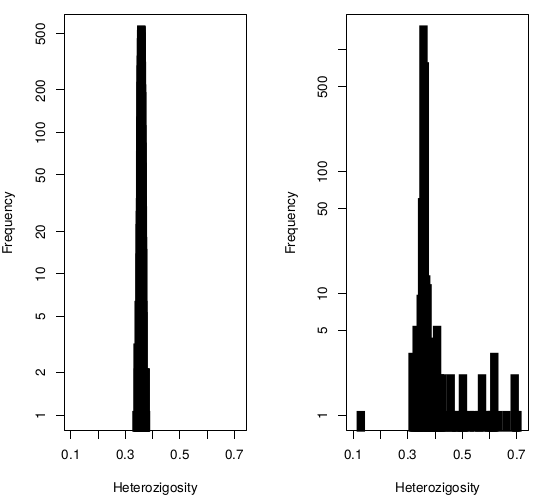
Exclusions
-
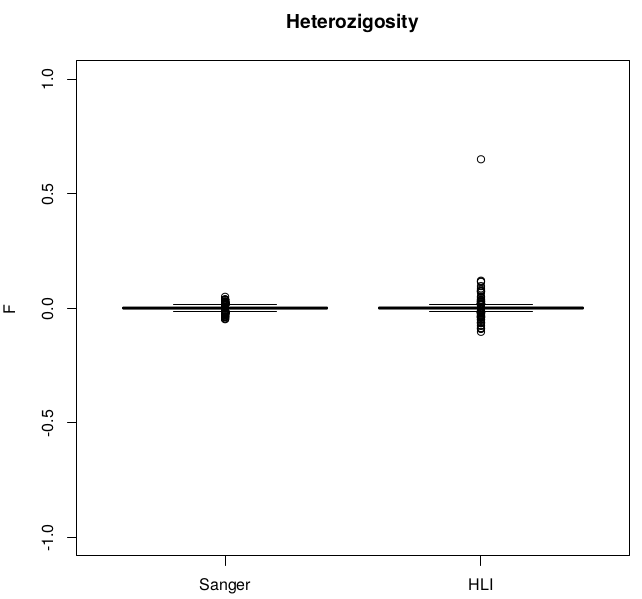
42 individuals excluded due to high (Het>0.4) heterozygosity :
- 5 Twin Pairs (10 individuals)
- 22 unrelated (unpaired twins)
- 10 parents
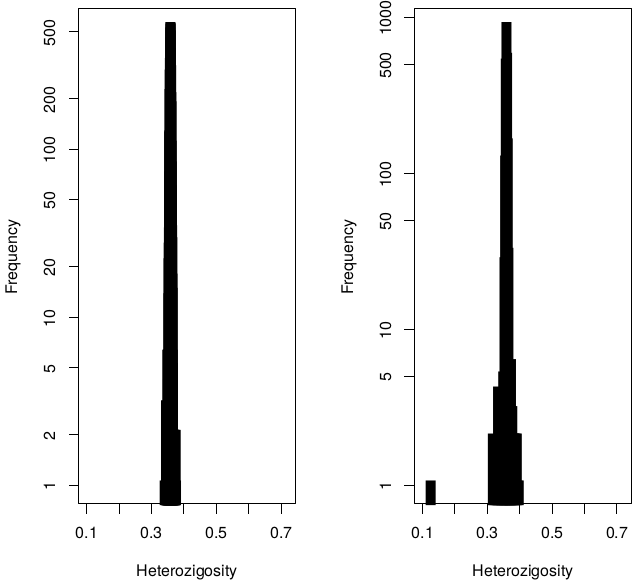
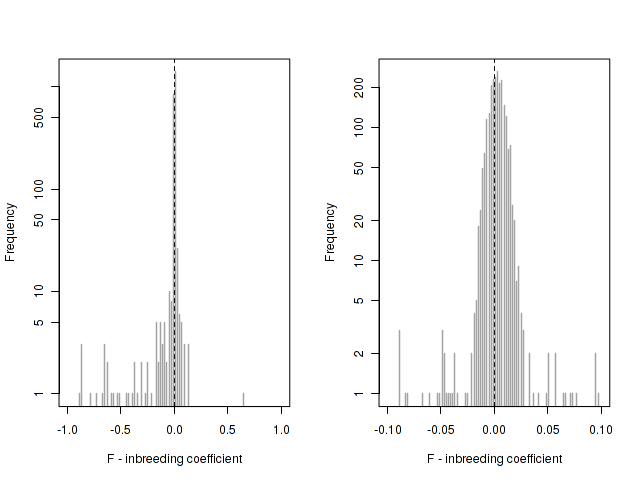
Heterozygosity before / after exclusions
Heterozigosity -- F
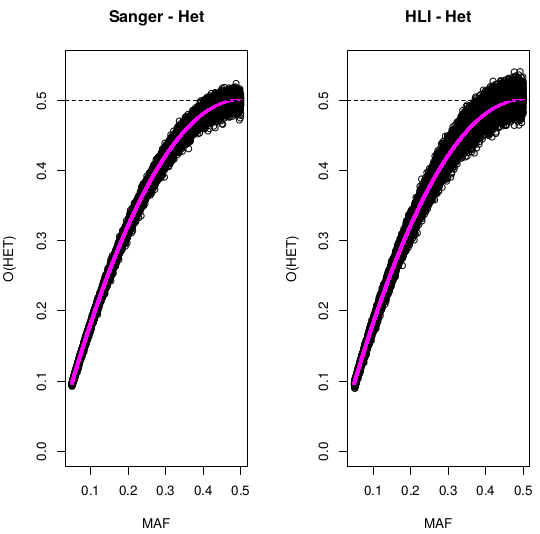
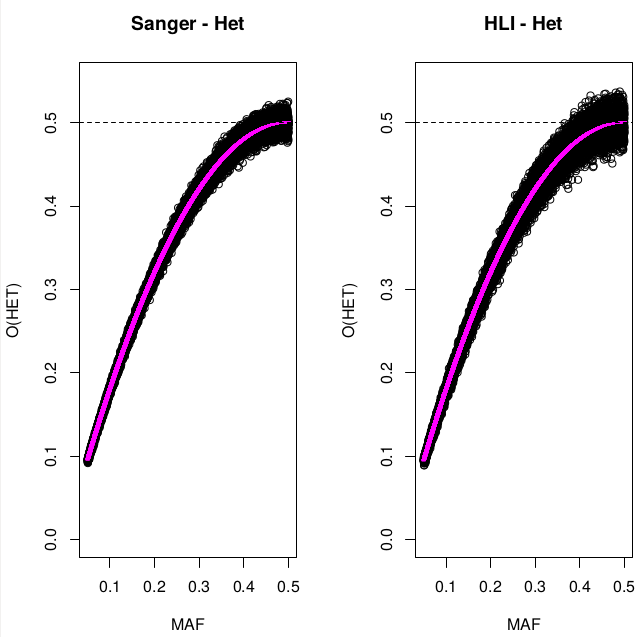
Heterozigosity O&E ~ MAF
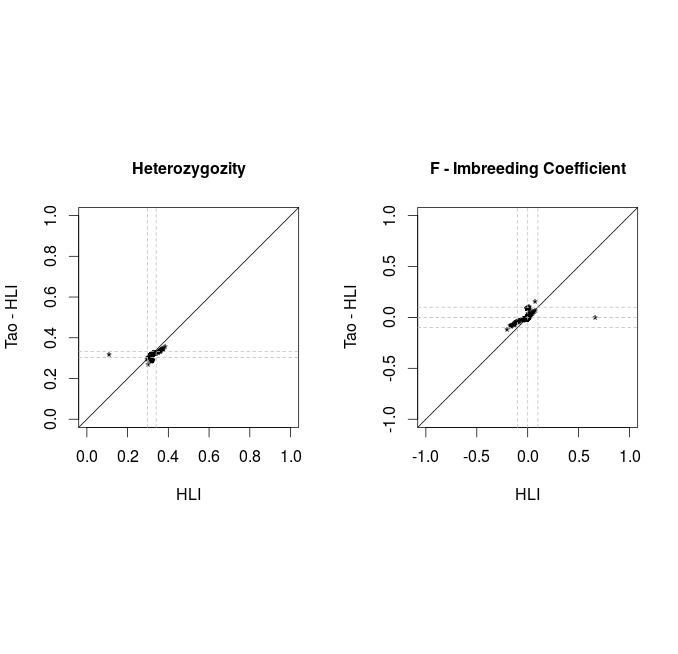
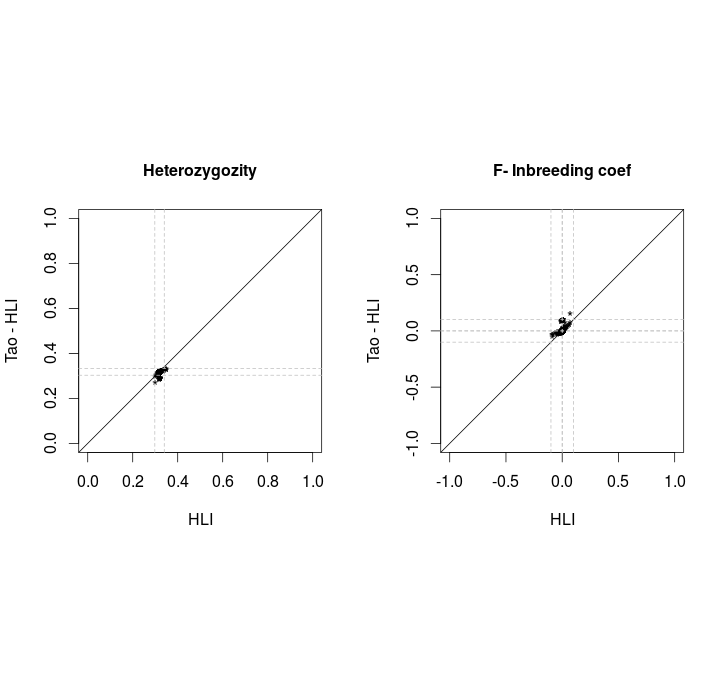
Releases comparison Hetererozygosity
Tao vs Alex
Only variants with MAF<0.05 and HWE<1E-6
Missing vcf file
[alvesa@athena HLI_HG38]$ ls | grep 6052
60521.vcf.gz
60521.vcf.gz.tbi
Client.Subject.ID Gender Ethnicity Birth.Date FamilyID Relation Zygosity Stool.Sample HLI.genome.ID 1731 60522 F White 10/4/35 762 Twin MZ TRUE 176500025 |
|
|
|
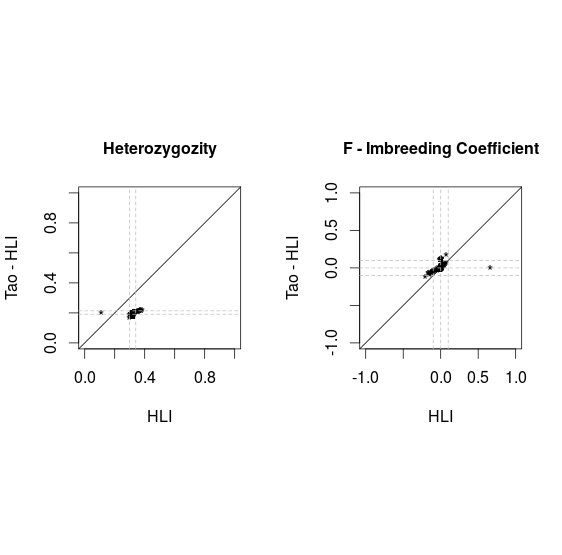
Comparison PLINK vs VCF
Comparison PLINK vs VCF
After removal of 16 individuals shared with Tao's release with |F|>.1
Comparison PLINK vs VCF
Before filtering SNP with MAF<0.05 and HWE<1E-6
Releases comparison Zygosity & IBD
Sanger vs Alex
Relatedness ~ Zygosity
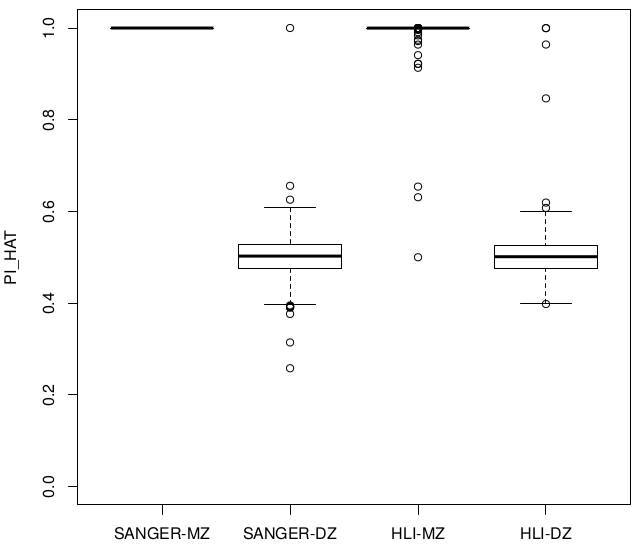
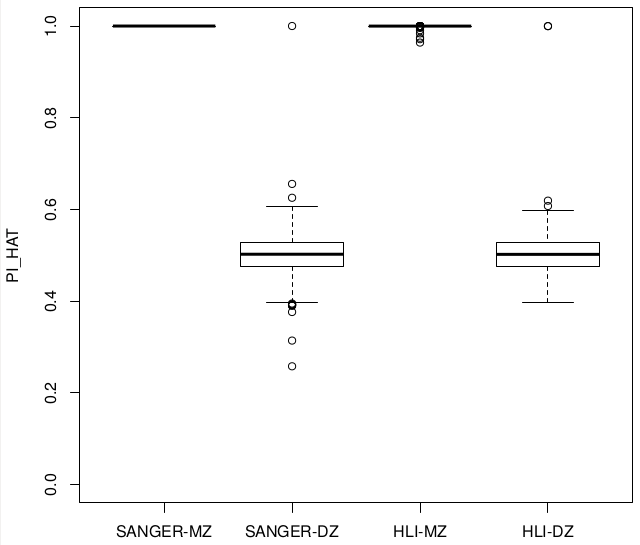
Concordance
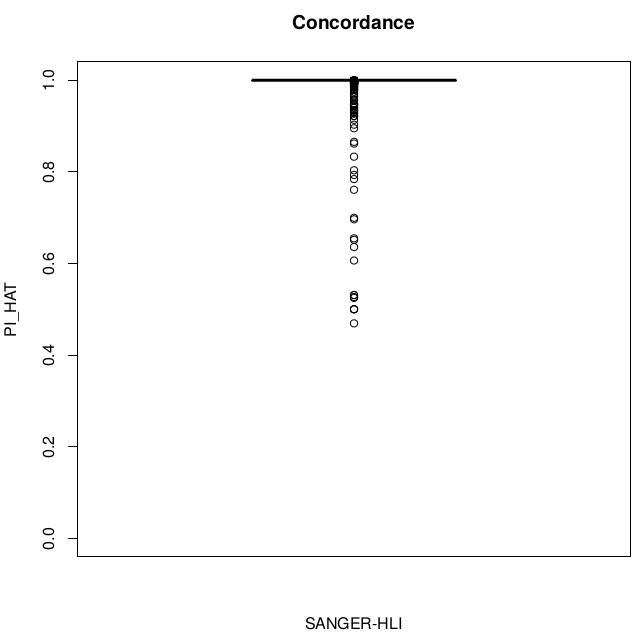
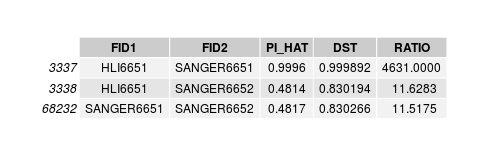
SUM(I(PI_HAT<0.9)) = 22
SUM(I(PI_HAT<0.9)) = 5
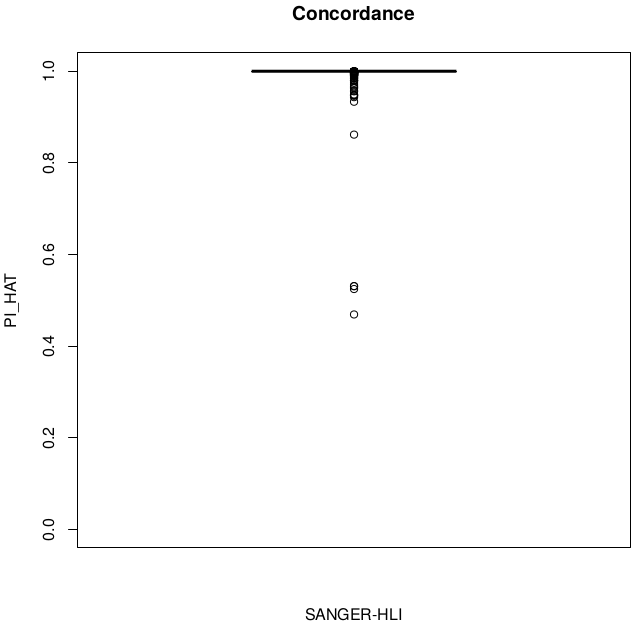
Discordant individuals
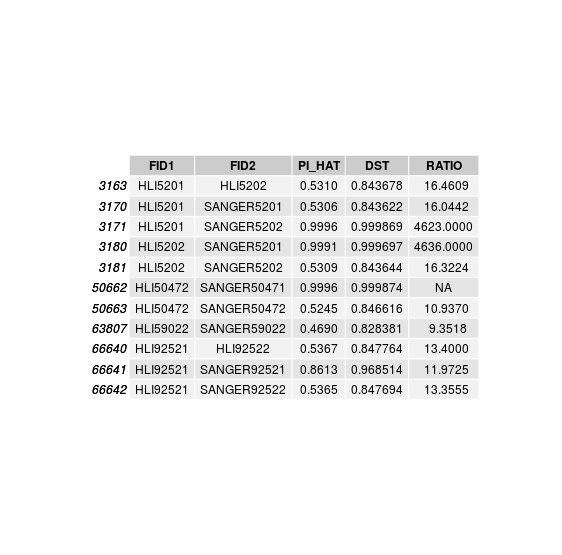
Discordant individuals: 5201, 5202, 50472, 59022, 92521
disconcordance analysis
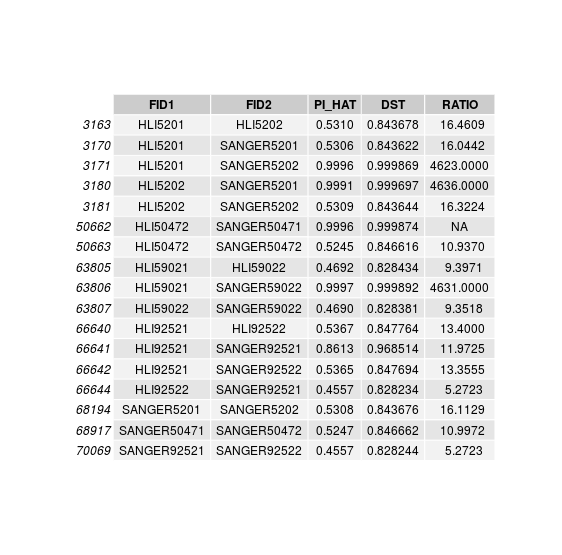
Discordant individuals: 5201, 5202, 50472, 59022, 92521
"Inbreed" individual
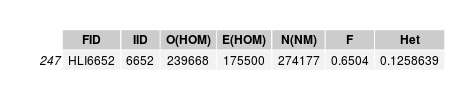
Individual is unrelated to any sequenced sample, including its chip-genotyped sample
Sample swaps
Sample swap within twin pairs:
HLI 5202 is actually SANGER 5201
HLI 5201 is actually SANGER 5202
Sample swaps in unpaired twins:
HLI 50472 is actually SANGER 50471
HLI 59021 is actually SANGER 59022
Individuals with low relatedness with SANGER sample:
HLI 92521 is matched to SANGER 92521 but with lower relatedness.
Unpaired twins either in SANGER of HLI:
SANGER59021
HLI50471
Conclusions
- 2357 samples analysed
- 1 missing individual as per Tao's release
- 47 individuals discarded with signs of contamination
- 2% of all individuals with |F|>0.1
- 4 sample swaps
Future work
- Deploy
- VCF and plink releases with highpass regions filtered in and problematic individuals filtered out
- merged VCF with all individuals and all regions
WG HLI HG38 VCF
GenotypeConcordance: VCFtools
Exon HG19 vcf
Coordinate change HG38:
crossmap
Convert PLINK to VCF
WGS data HG38 VCF
(HLI)
Merge Datasets
Array data PLINK HG18 (Sanger merge)
Coordinate change HG38:
crossmap
Flipping alleles from positive to reference strand
Convert VCF to PLINK
Convert VCF to PLINK
IBS Analysis
Filter in MAF>5% HWE>1E-6
Merge VCF: BCFtools
WG HLI HG38 VCF
Convert to plink: BCFtools
flip +ve strand: plink
Annotate VCF: GATK
Filter highpass regions: Bedtools
gen file
matrixeqtl file
phasing: shapeit2
convert files: qctool / gtools
plink file
phased plink file
QC individuals
EG Meeting -- HLI WGS HG38
By acoutoal



