Introduction to epigenetics and ChIP-Seq
Aleksandra Galitsyna
“Analysis of omics data” course
Skoltech Term 4
20 April 2020

This presentation can be found at https://slides.com/agalicina/epigenetics-practice1-2020
Introduction: sequencing
"Computational analysis of
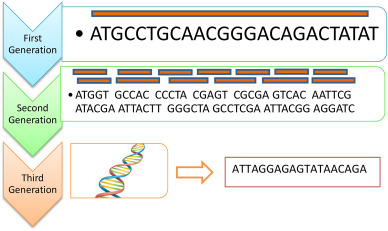
Sanger
Illumina, Roche 454, SOLiD
Nanopore
Illumina sequencing
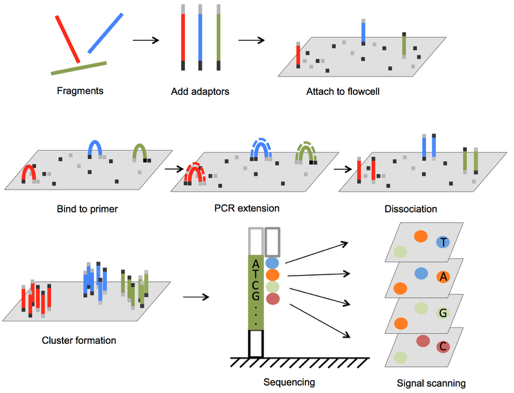
Illumina sequencers


NGS techniques diversity

Epigenetics
Wasserman & Sandelin, 2004
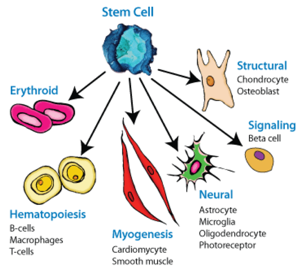
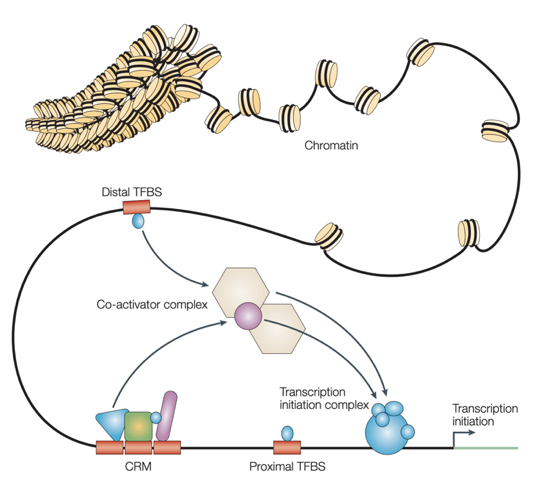
Sequencing for epigenetics
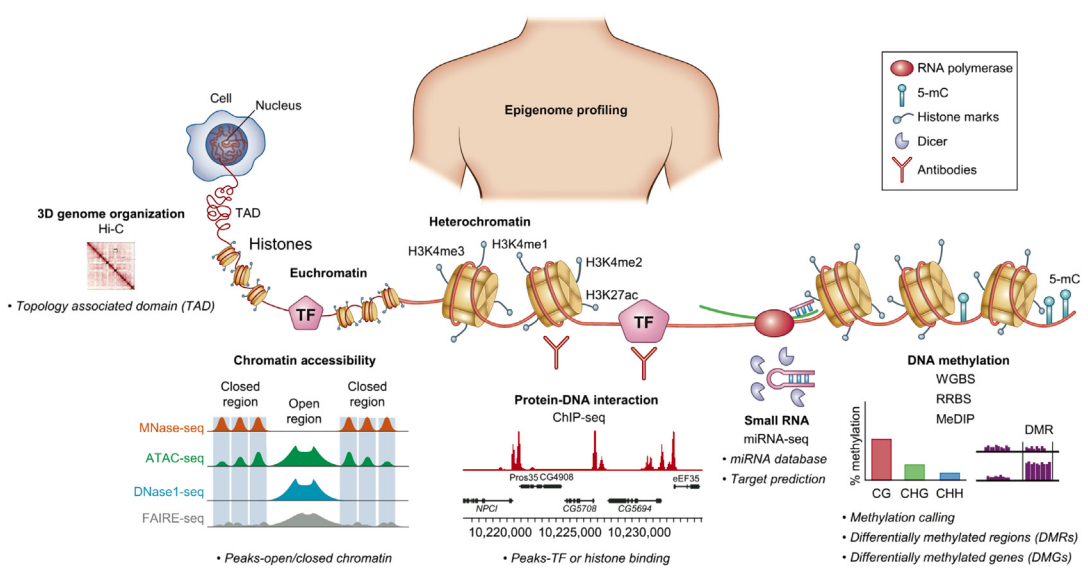
ChIP-Seq: experimental protocol
- Aim: find all regions in the genome bound by a specic transcription factor (or bearing a specic histone modication)
- Answer: ChIP-Seq, Chromatin immunoprecipitation with sequencing
Park 2009
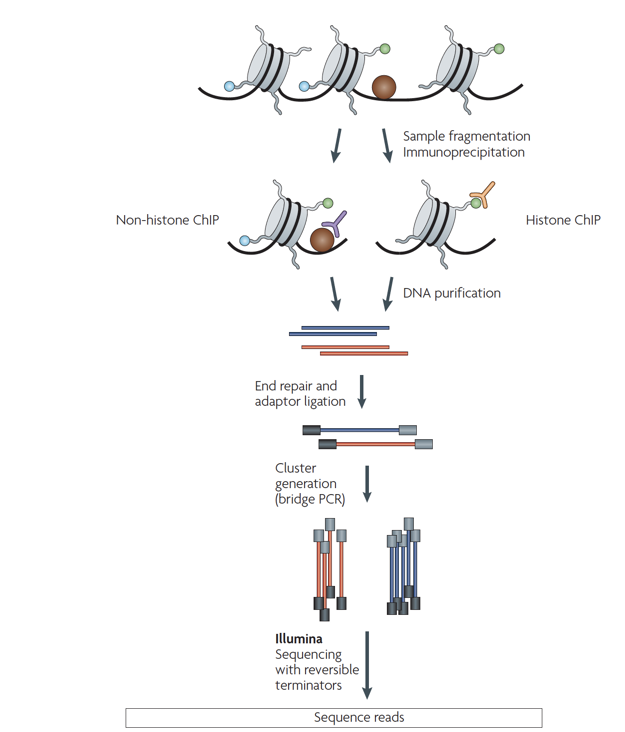
ChIP-Seq: density of DNA fragments
- We expect to find different results for plus (Watson) and minus (Crick) strands of DNA:
Park 2009
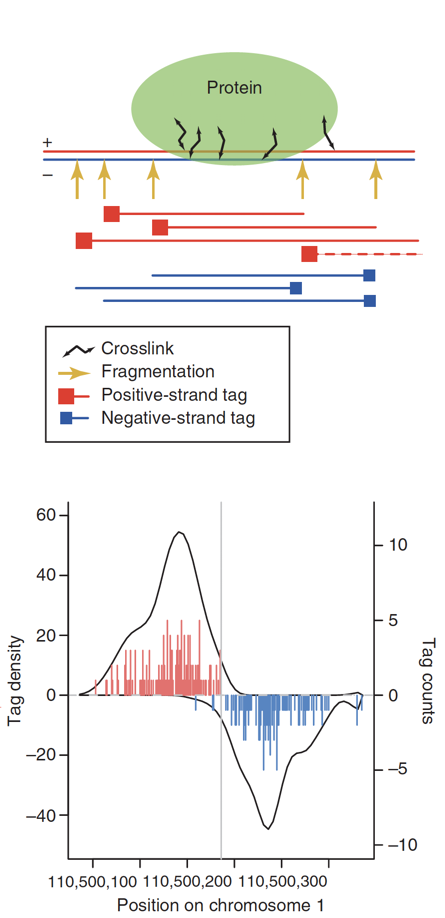
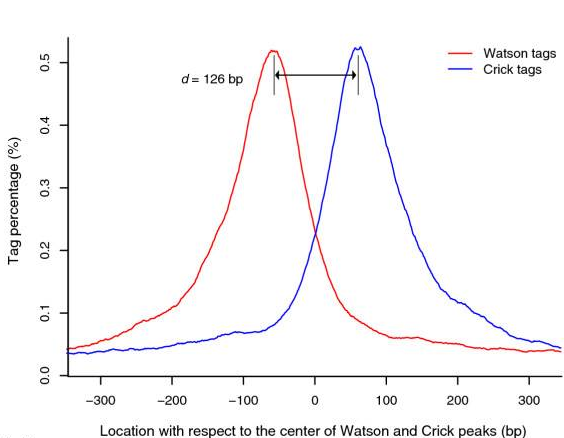
ChIP-Seq: Cross-Correlation
- We expect to find different results for plus (Watson) and minus (Crick) strands of DNA:
Park 2009
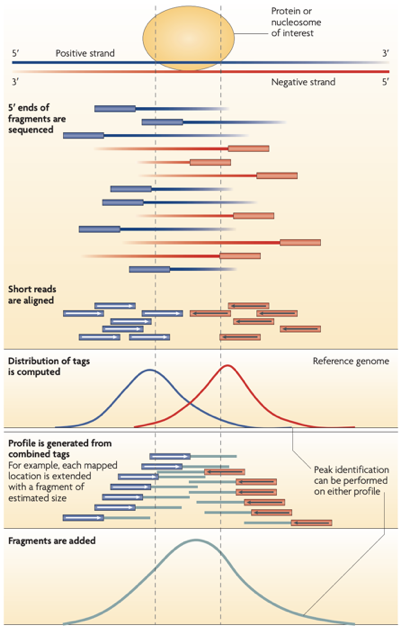
ChIP-Seq: Cross-Correlation
-
The cross-correlation typically peaks at the shift corresponding to the fragment length and the shift corresponding to the read length.
-
NSC (normalized strand cross-correlation coefficient):
The ratio between the cross-correlation at the fragment length and the background crosscorrelation. -
RSC (relative strand cross-correlation coefficient):
The ratio between cross-correlation at the fragment length and the crosscorrelation at the read length. -
Very successful ChIP experiments generally have NSC>1.05 and RSC>0.8
Bailey 2013

Input experiment
- ChIP-Seq control without immunoprecipitation step, used for normalisation of ChIP-Seq.
-
Unwanted factors contributing to sequencing & processing output:
- DNA-shearing is not uniform across genome -> more reads in open chromatin regions,
- Amplification bias (GC content),
- Repetitive regions,
- DNA accessibility
Park 2009
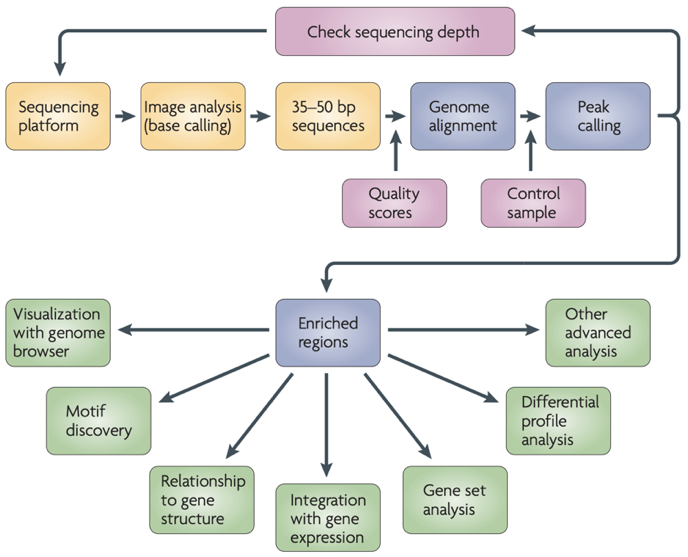
ChIP-Seq data processing
Park 2009
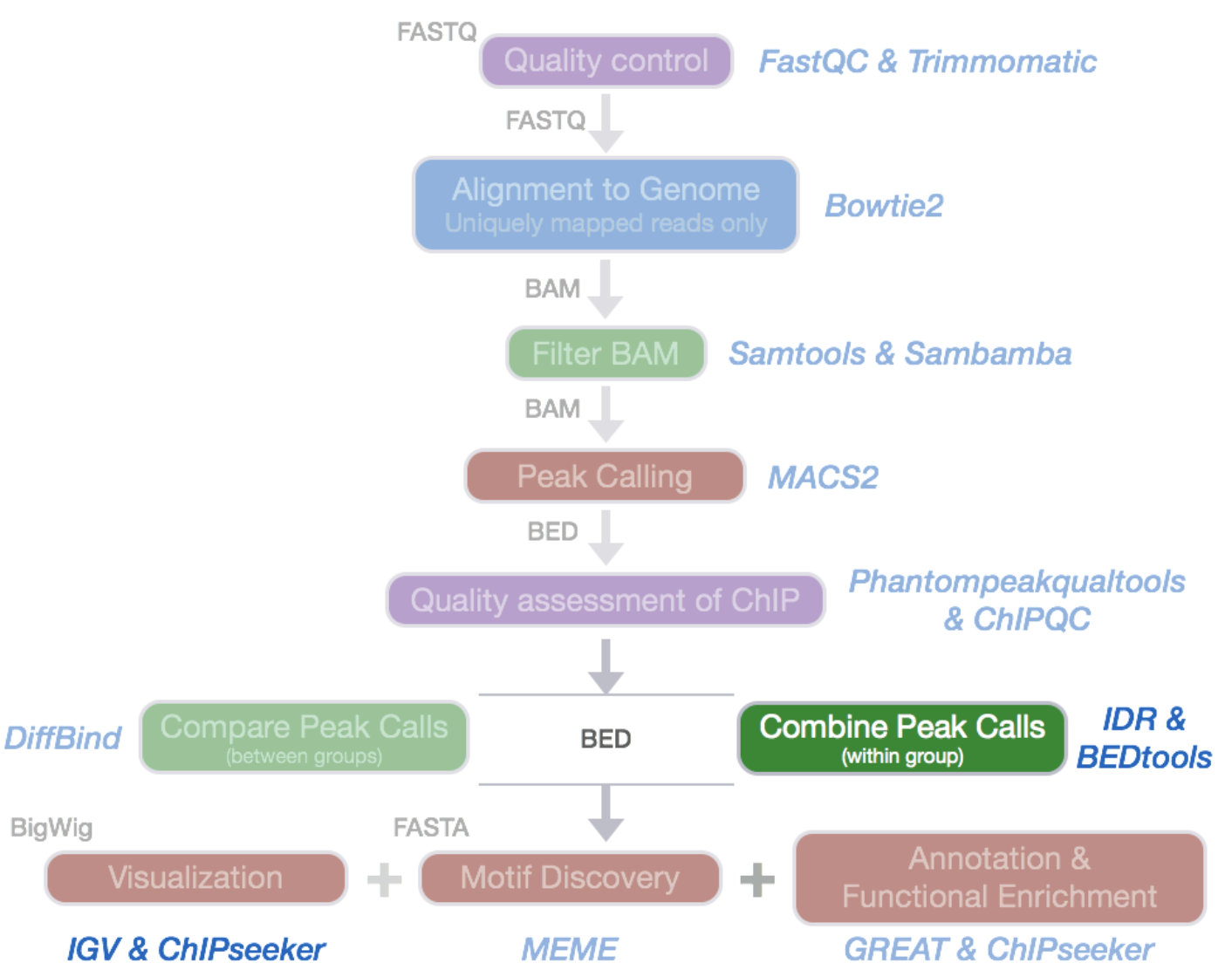
ChIP-Seq data processing: implementation
ENCODE Standards:
https://www.encodeproject.org/chip-seq/transcription_factor/
ChIP-Seq peak calling
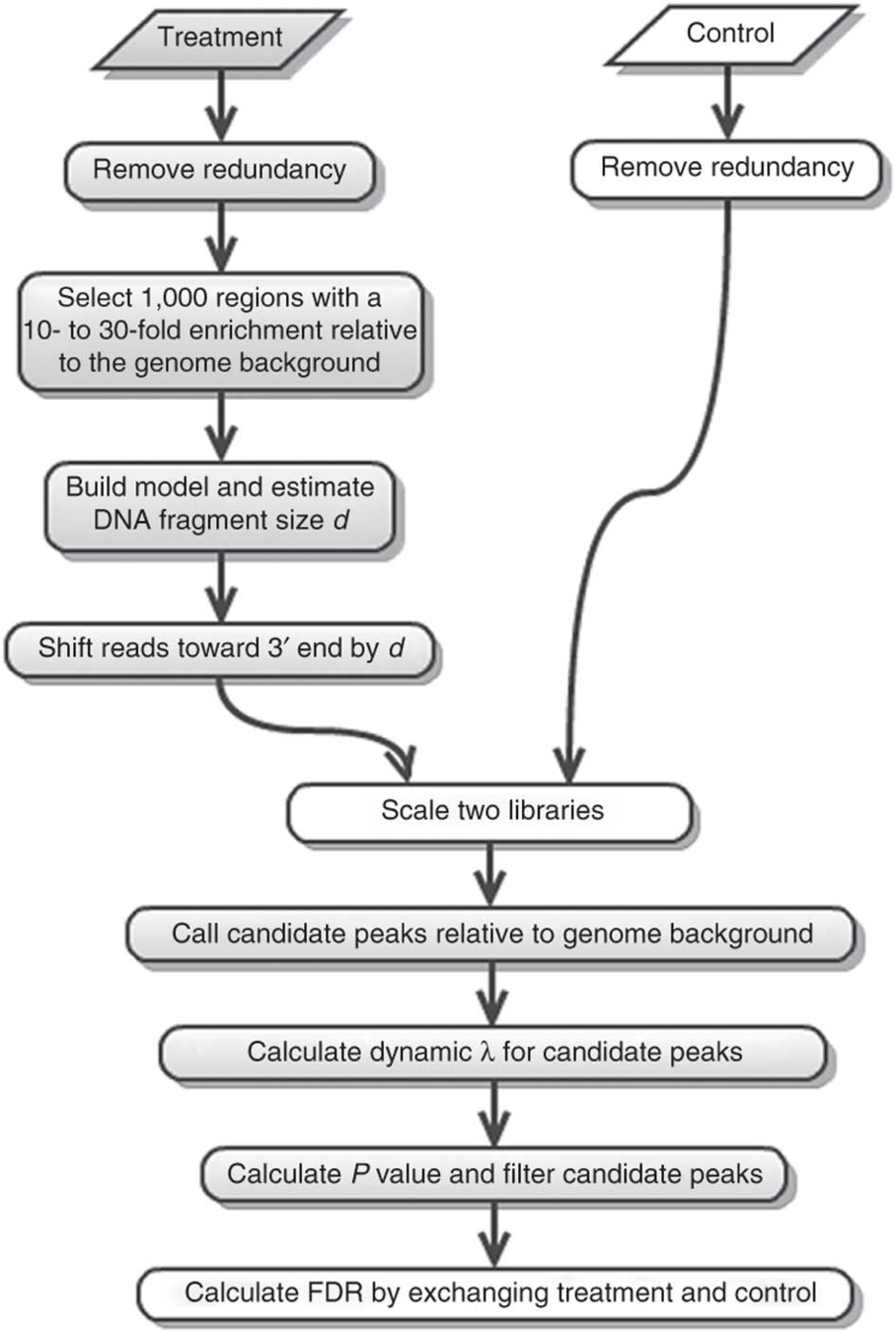
MACS2 is the most common software:
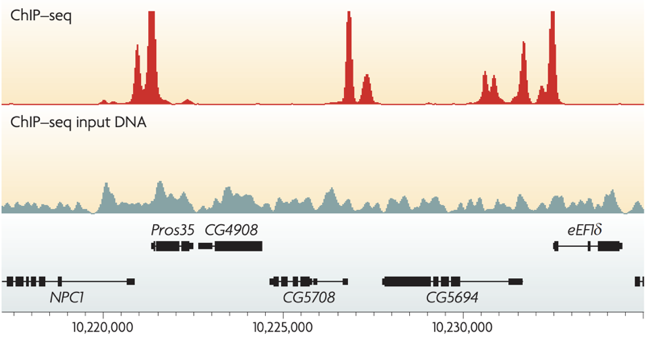
ChIP-Seq peak calling result

Binding of protein is typically specific to DNA sequence:
DNase-Seq: experimental protocol
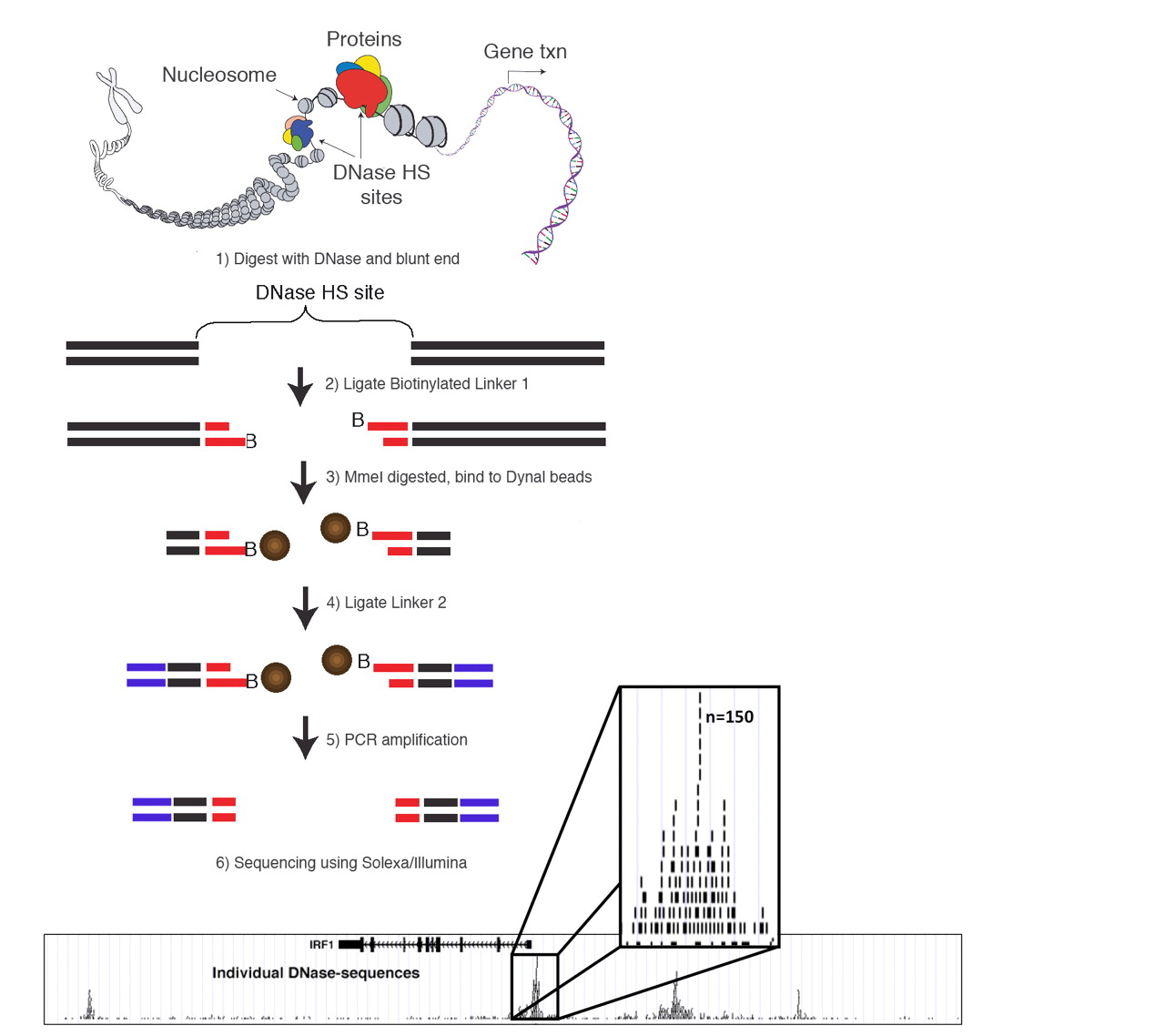
- DNase I hypersensitive sites sequencing
ATAC-Seq: experimental protocol
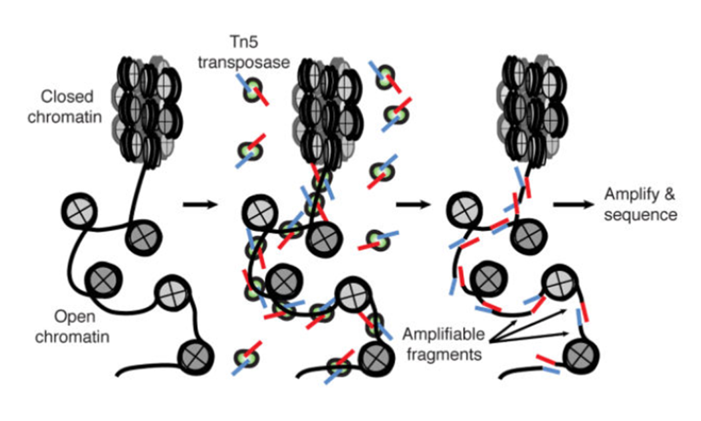
- Assay for Transposase-Accessible Chromatin with high-throughput sequencing
Nucleosome-free regions detection method- Tagmentation: Tn5 transposase cuts DNA and ligates adaptors to open DNA
Buenrostro et al., 2013
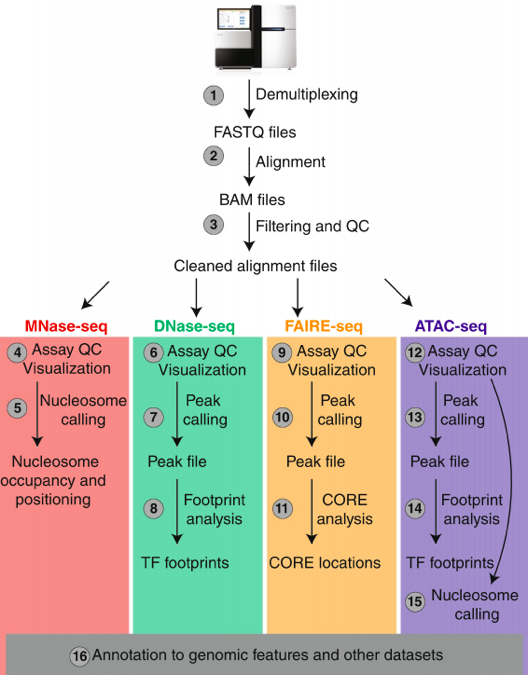
"Block-schemes" of Seqs:
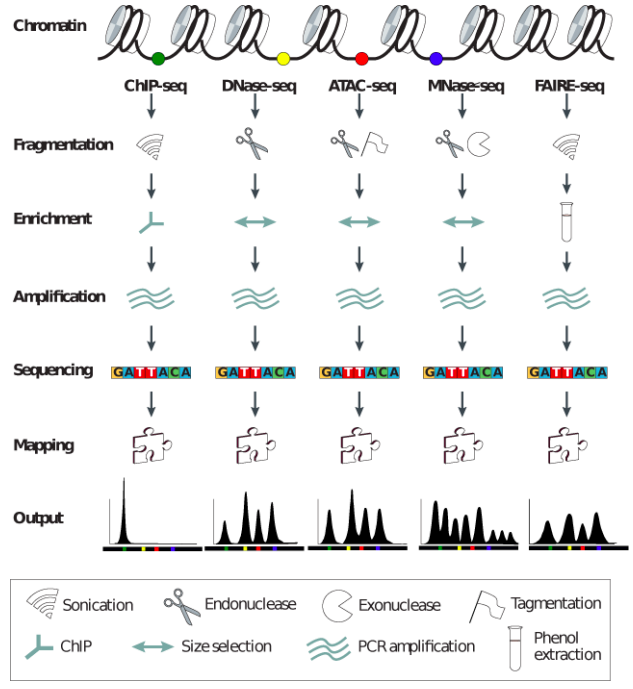
Meyer & Liu Nat. Rev. Gen. 2014
Data processing of Seqs:
Meyer & Liu Nat. Rev. Gen. 2014
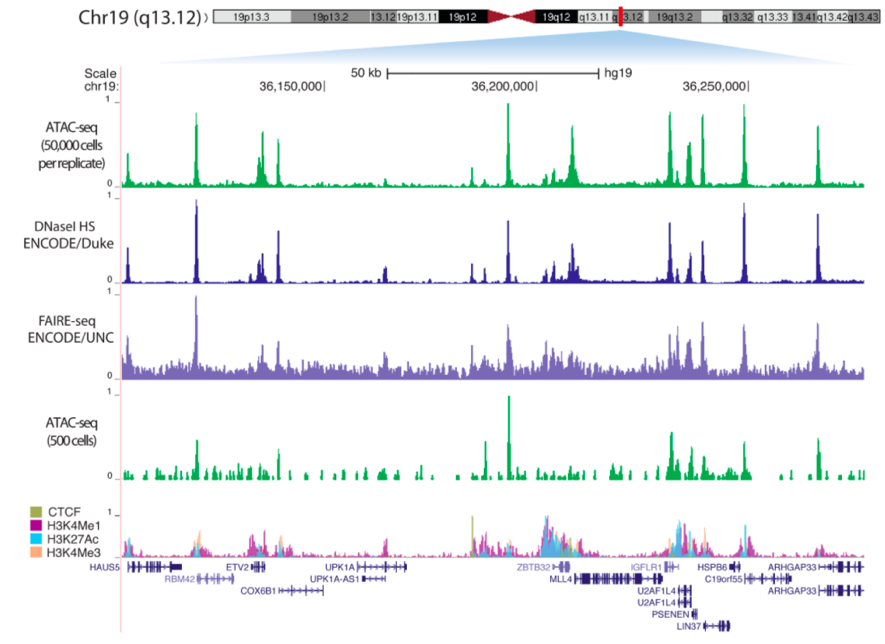
Examples of tracks:
Meyer & Liu Nat. Rev. Gen. 2014
In theory:

Methylation of basepairs:


BS-Seq: protocol
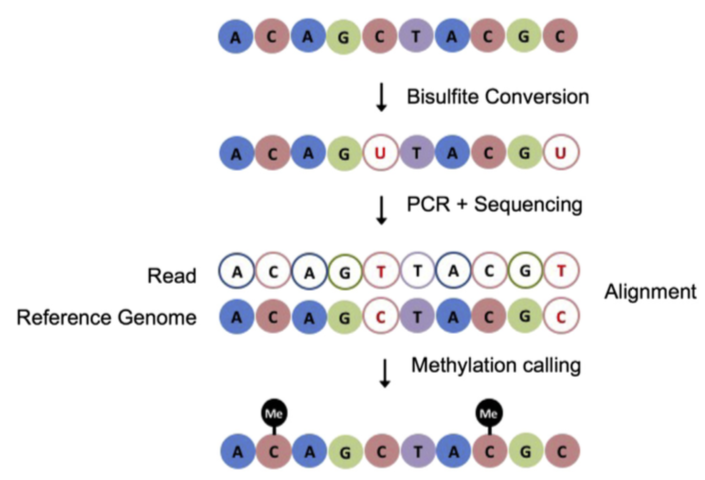
Bisulfite-Seq
BS-Seq: data processing
Bisulfite-Seq
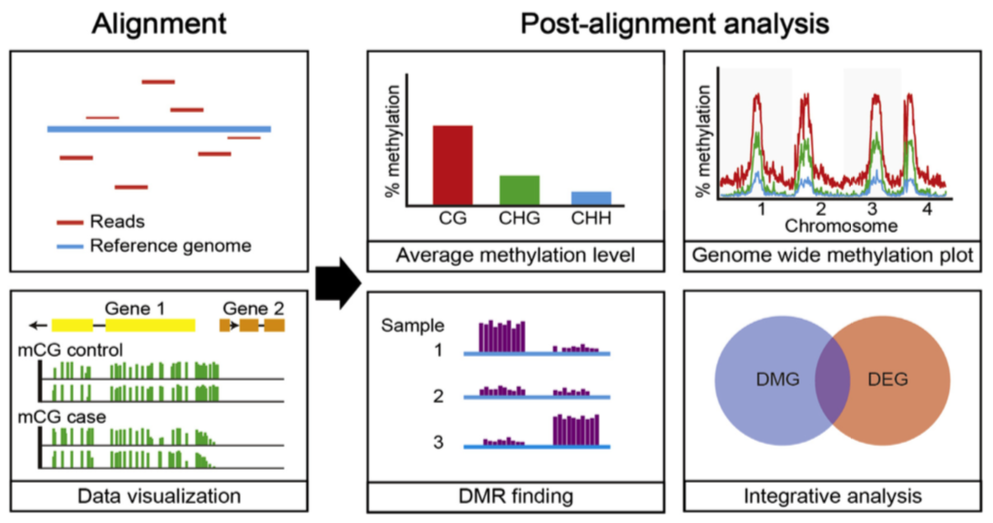
Bismark: methylation caller for Bisulfite-Seq
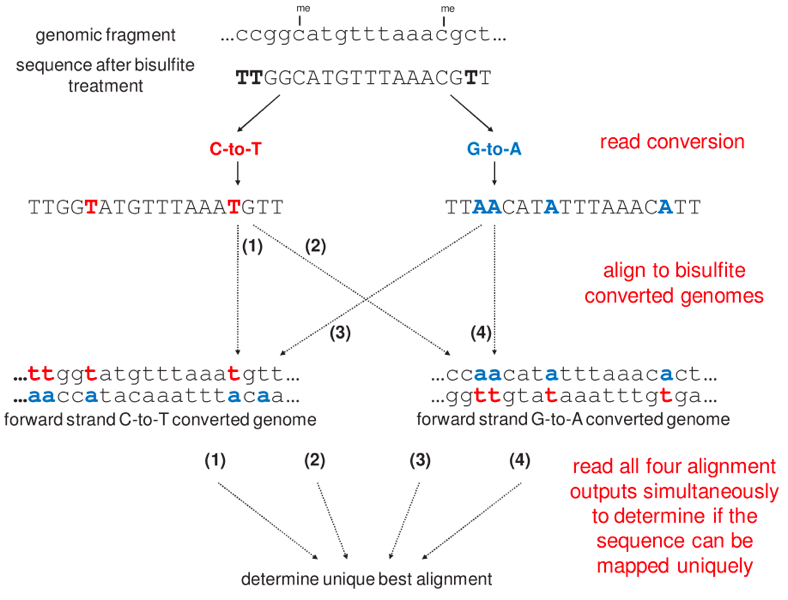
Before we start the practice...
- 5 practices on NGS for epigenetics in a row
- Each practice = a hometask with a report.
- Deadline is within a week.
- The report tasks, requirements and number of points are explicitly specified in the presentation in red
- You can have 5 points max per this hometask. Extra points for great answers might compensate for incomplete or erroneous tasks.
Before we start the practice...
- We will work in browser and with terminal. We also need to copy files from the remote server, so install appropriate software, if needed (your favourite terminal distributive,
Putty: https://www.putty.org/ or WinSCP: https://winscp.net/eng/download.php) - File formats are denoted in CAPS in the presentation: FASTQ, BED, SAM etc.
- Code is running in white boxes with monospace font, try to use the copy button, but be careful:
some file names are surrounded by "<>". This indicates that it should be replaced by the name of your file.
echo "Bioinformatics has never been easier! Copy&paste wisely, or rm -rf ~/ will strike you one day."
Before we start the practice...
Review of some useful bash commands:
- List files in the current directory:
- Check the current position in the directory tree:
- Open text file for reading:
- Run command help, one of the options:
(some of them work for ones
but not the others)
- Copy file:
- Enter directory:
- Exit the directory:
- Create directory:
- File decompression:
lspwdless file.txtsome_command -hsome_command --helpman commandcp target_file destination_foldercd directory_name/cd ../ls -htsmkdirgzip -d file.gzunzip file.zipGet in touch with your data: step 1
1. Login to the server via terminal:
or use Putty on Windows.
(example instructions https://www.ssh.com/ssh/putty/windows/#sec-Configuration-options-and-saved-profiles)
2. Create the folder with your project:
3. Activate prepared environment:
(if you have problems with it, please, contact the TA)
4. Check your environment:
ssh username@servernamemkdir EpiPract1
cd EpiPract1export PATH="/home/galitsyna/anaconda3/bin:$PATH"
printf "envs_dirs:\n - /home/galitsyna/anaconda3/envs/" > .condarc
conda activate epigeneticsls # Should list all the filesin the directory
pwd # Should result in your currentl location
conda list # Should list all the programs installed, if conda imported successfully.Get in touch with your data: step 2
- Find your datasets (here or in the table on the right):
- Copy to the local folder:
- Check the content and file sizes:
less /home/galitsyna/EpiPract1/students_spreadsheet.txtcp /home/galitsyna/EpiPract1/FILES/<your_file.fa.gz> ~/EpiPract1/
mkdir ~/EpiPract1/GENOME/
cp /home/galitsyna/EpiPract1/GENOME/chr[5,6].fa ~/EpiPract1/GENOME/
cp /home/galitsyna/EpiPract1/GENOME/chrom_sizes.tsv ~/EpiPract1/GENOME/ls -hts
ls -hts GENOME/You should have:
~/EpiPract1/ directory:
- FASTQ file (<your_file.fa.gz>) from the list on the right
~/EpiPract1/GENOME/ directory:
- FASTA files with hg38 reference genome
for chr5, chr6 (chr5.fa, chr6.fa)
- TSV (Tab Separated Values) file with
chromosome sizes (chrom_sizes.tsv)
| Name | File |
|---|---|
| Anastasia Pivnyuk | ENCFF001CWQ_1.fa.gz |
| Nikita Sharaev | ENCFF001CWQ_2.fa.gz |
| Artemy Shumskiy | ENCFF002ERE_1.fa.gz |
| Dmitrii Kriukov | ENCFF002ERE_2.fa.gz |
| Pletenev Ilya | ENCFF228WDF_1.fa.gz |
| Konstantin Chernyshov | ENCFF228WDF_2.fa.gz |
| Ivan Kuznetsov | ENCFF280UJR_1.fa.gz |
| Anna Kalinina | ENCFF280UJR_2.fa.gz |
| Sofya Kasatskaya | ENCFF288DXQ_1.fa.gz |
| Julia Bocharkina | ENCFF288DXQ_2.fa.gz |
| Vasily Borodin | ENCFF291MFN_1.fa.gz |
| Sofia Kamalyan | ENCFF291MFN_2.fa.gz |
| Anna Krasivskaya | ENCFF512FUR_1.fa.gz |
| Aleksandra Ozerova | ENCFF512FUR_2.fa.gz |
| Victoria Kobets | ENCFF586YZS_1.fa.gz |
| Slesareva Anastasiia | ENCFF586YZS_2.fa.gz |
| Mikhail Moldovan | ENCFF591XCX_1.fa.gz |
| Viktor Mamontov | ENCFF591XCX_2.fa.gz |
| Evgeniia Alekseeva | ENCFF601JYM_1.fa.gz |
| Trofimova Anna | ENCFF601JYM_2.fa.gz |
Get in touch with your data: step 3
- Open FASTQ files, check the format
(see also: http://emea.support.illumina.com/bulletins/2016/04/fastq-files-explained.html)
- Check FASTQ files, what is the number of rows vs the number of entries?
- Run fastqc (a tool for FASTQ quality control, or QC)
gzip -d <file.fastq.gz> # This will result in <file.fastq> unzipped file
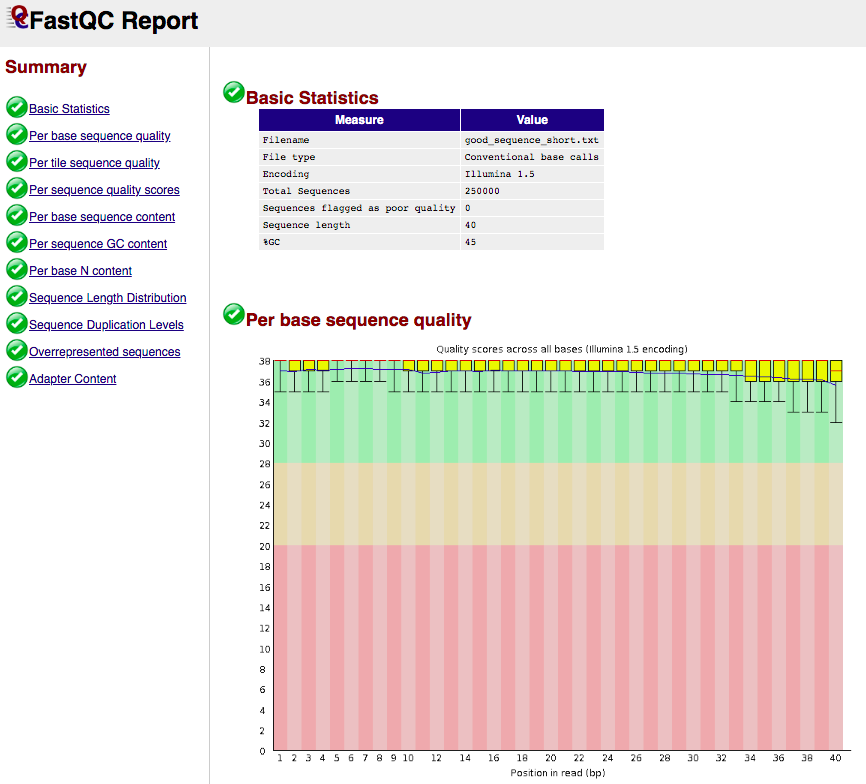
less <file.fastq>wc -l <file.fastq>fastqc <file.fastq>Task 1 (0.5 points): report the number of entries in your FASTQ file, read length and passed/failed fastqc filters. Provide a brief explanation.
Task 2 (1 points): go to ENCODE (https://www.encodeproject.org/). Find ID of your file (e.g. ENCFF601JYM) there. Report the experiment type, sequencing platform, cell type (Biosample summary) for your NGS data.
NGS data typical workflow example
Let's review the NGS data processing workflow:
- Quality control: fastqc on initial FASTQ
- Index build: bowtie2-build on initial chromosomes files
- Read mapping: FASTQ → SAM/BAM
- Filtering of mapped reads: samtools on SAM from p.3
- Conversion to BED: bedtools on SAM from p.4
FASTQC report example
- Takes FASTQ as input:
- Output format: html and plain text
fastqc <input.fastq>ls
# input.fastq input_fastqc/
cd input_fastqc/
ls
# fastqc_data.txt fastqc_report.html Icons Images summary.txtDon't run this code, it's FYI
FASTQC report example
- Example html output:
(see https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ for more examples)
Genome index build example
(Manual: http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml)
- Required for fast search of query sequence during mapping
- Takes fasta with reference as input (comma-separated list) and prefix name.
- Example run and resulting database:
bowtie2-build -h
bowtie2-build chr1.fa,chr2.fa,chr3.fa,chr4.fa hg38
# Check the results:
ls -hs
# 938M hg38.1.bt2 701M hg38.2.bt2 12K hg38.3.bt2 701M hg38.4.bt2 3.0G hg38.fa 938M hg38.rev.1.bt2 701M hg38.rev.2.bt2
bowtie2-inspect -n hg38Don't run this code, it's FYI
Read mapping example
- Takes query FASTQ and indexed database as input.
- Output is in SAM or BAM format
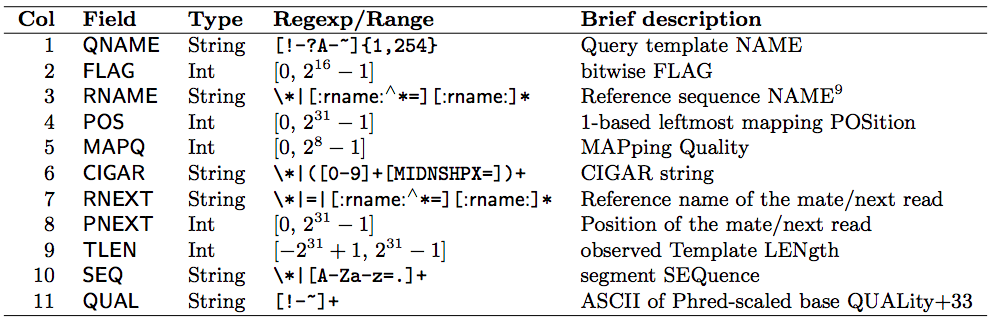
(SAM format specification: https://samtools.github.io/hts-specs/SAMv1.pdf) - SAM is a TSV file with header, BAM is its compressed binary version. The columns are:
Read mapping example
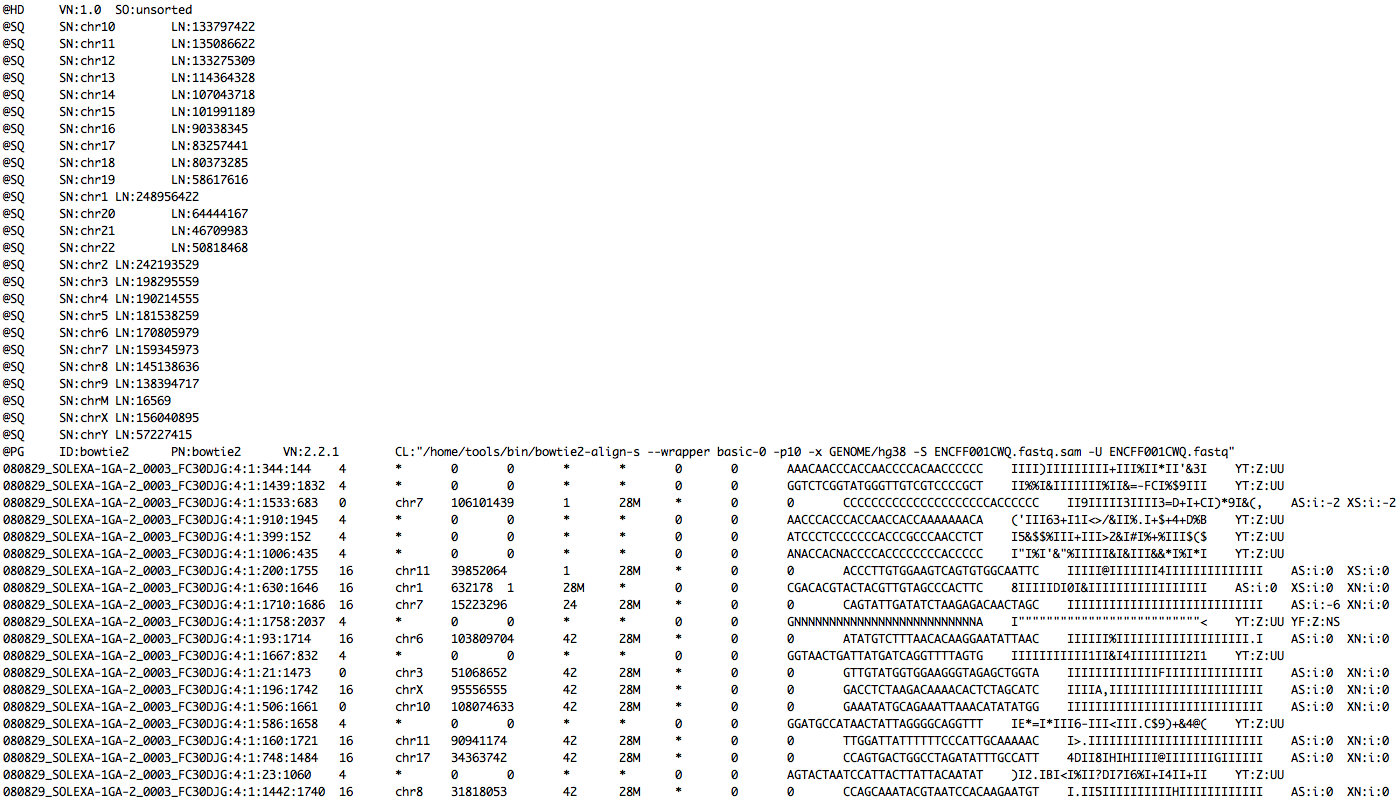
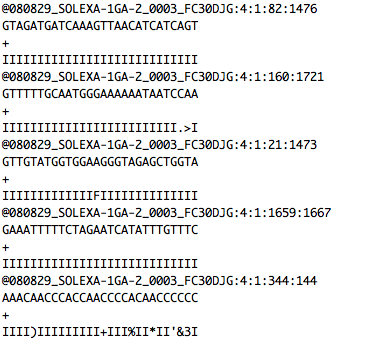
FASTQ
+ indexed database

-> SAM file
Samtools example
(Manual: http://www.htslib.org/doc/samtools.html)
- A toolkit designed for SAM/BAM manipulations
- View:
- Statistics assessment:
- Format conversion:
- Filtering:
samtools view file.samsamtools view -F 4 input.sam
samtools stats file.samsamtools view -b file.sam > file.bamBedtools example
(Manual: https://bedtools.readthedocs.io/en/latest/content/bedtools-suite.html
BED format specification: https://genome.ucsc.edu/FAQ/FAQformat.html)
- A toolkit for conversion and manipulations with BED:
- A powerful tool for genomic arithmetics:
(Tutorial: https://bedtools.readthedocs.io/en/latest/)
bedtools bamtobed -i file.bam > file.bed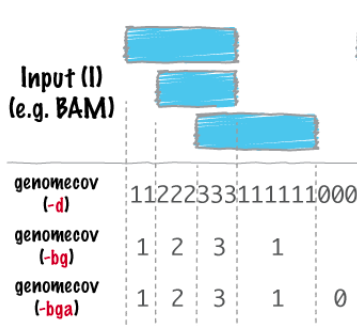
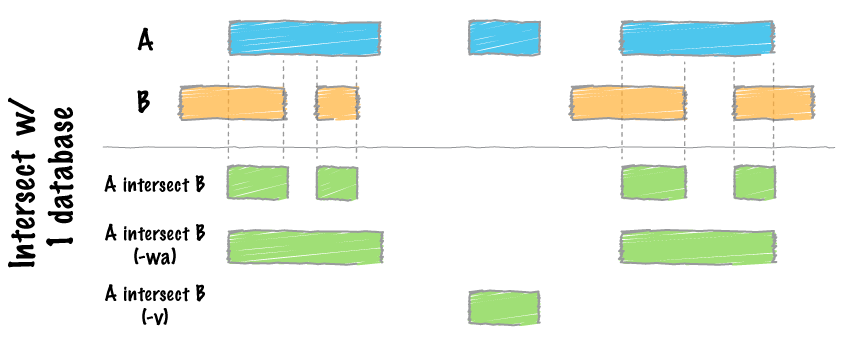
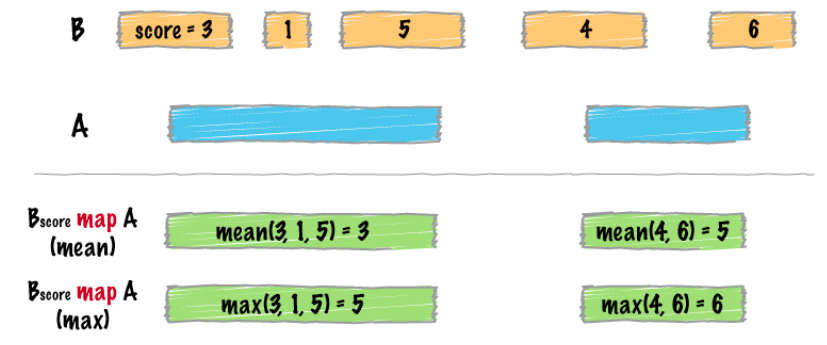
map
intersect
Other formats
- BED
- BEDGRAPH
- BIGWIG: binary compressed format, created with UCSC tools (see below)
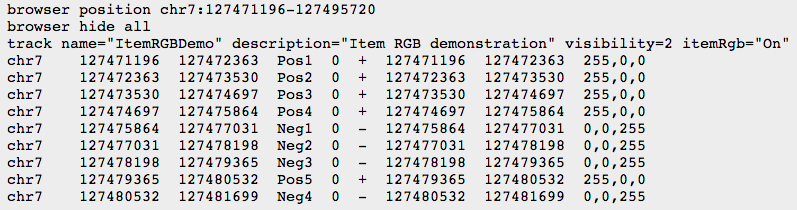
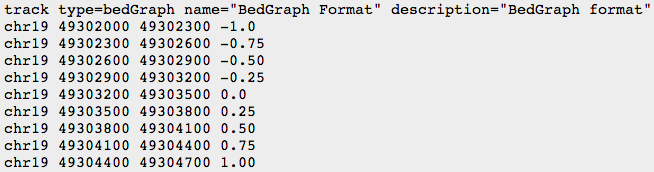
UCSC genome browser & UCSC tools
- UCSC maintains genome browser and has a set of tools: https://genome-euro.ucsc.edu/

UCSC tools example
Full set of compiled tools: http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
- We will need file conversion, check the documentation:
bedGraphToBigWig -hNext step: index build and mapping
These steps are time-consuming, but we have to complete them:
- Build genome index with name prefix hg38_chr5-6_index:
- Run mapping of your FASTQ file for the index hg38_chr5-6_index:
Task 3 (1 points): report the number of unique, multiple mappings and unmapped reads in your FASTQ file. Report the major chromosome, on which your reads have mapped.
cd GENOME/
bowtie2-build chr5.fa,chr6.fa hg38_chr5-6_indexbowtie2-build -hbowtie2 -hbowtie2 --very-sensitive -x ./GENOME/hg38_chr5-6_index -U <file.fastq> -S <file.sam>less <file.sam>ls
cd ../Filtering
- Filter only uniquely mapped reads with high alignment score, sort resulting file:
samtools view -h -F 4 <file.sam> | lesssamtools --helpsamtools view -h -q 10 <file.sam> > <file_filtered.sam>samtools view -b -q 10 <file.sam> > <file_filtered.bam>
samtools sort <file_filtered.bam> > <file_filtered.sorted.bam>File format conversion
- Read the documentation of bedtools:
- Convert BAM to BEDGRAPH genome coverage:
- Create files with genomic windows (1Kbp bin size, don't forget to use your chromosome!). Convert BAM to BEDGRAPH coverage of windows:
bedtools makewindows -g GENOME/chrom_sizes.tsv -w 1000 > hg38_windows.1000.bed
grep "chr5" hg38_windows.1000.bed > chr5_windows.1000.bed
bedtools genomecov -ibam <file_filtered.sorted.bam> -bga -split > <file.genomecov.bedgraph>bedtools genomecov -h
bedtools makewindows -h
bedtools coverage -h
bedtools coverage -counts -a chr5_windows.1000.bed -b <file_filtered.sorted.bam> > <file.binned.bedgraph>- Check the resulting files, you should see genome coverage
files (*.genomecov.*) and binned datasets (*.binned.*):
ls
less <file.genomecov.bedgraph>
less <file.binned.bedgraph>BEDGRAPH tracks visualization
We will use UCSC browser for bedgraph files visualization.
- Download both files file.genomecov.bedgraph and file.binned.bedgraph to your local computer.
- Manually add the header to your file:
(see UCSC recommendations: https://genome.ucsc.edu/goldenPath/help/bedgraph.html)
track type=bedGraph visibility=full
- Go to UCSC genome browser, hg38 genome:
https://genome-euro.ucsc.edu/cgi-bin/hgTracks?db=hg38
UCSC genome browser
- Find "add custom tracks button".
- Check the upload form:
- Upload your modified BEDGRAPH files (with headers) to UCSC ("Choose file" -> "Submit").
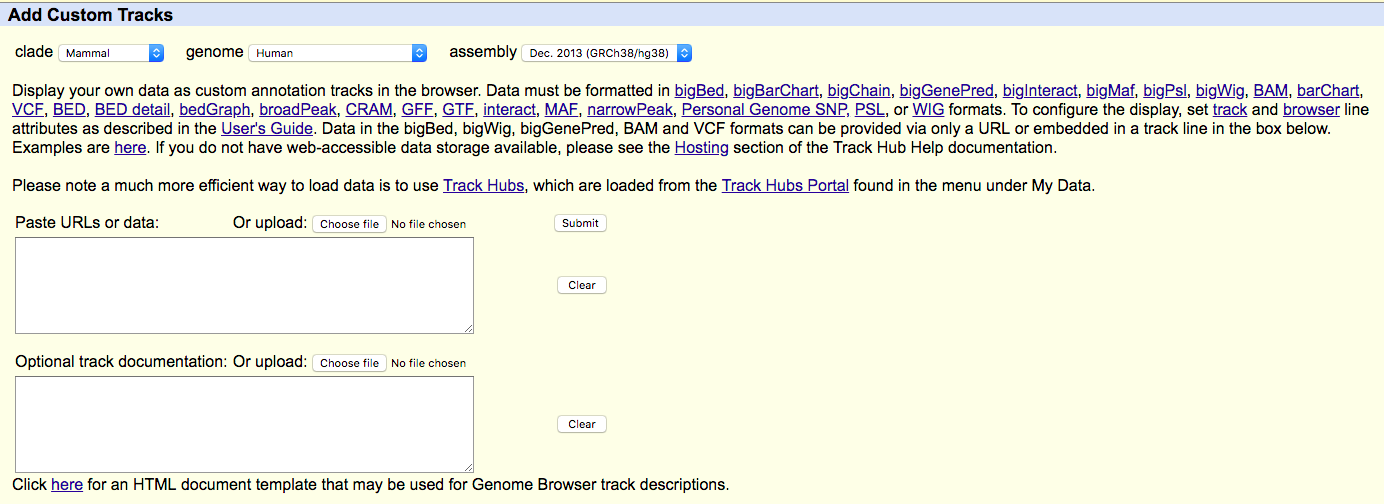
UCSC genome browser
Task 4 (1 point): Load both BEDGRAPH files with your dataset.
Select some gene from your chromosome and send the screenshot of 1 Kb, 10Kb and 1 Mb regions around it with visualization of your dataset.
Task 5 (1 points): Provide explanation of your observations. What do you expect from this type of experiment? What is the difference between genome coverage and window coverage?
Experiments comparison
- Convert BEDGRAPH to BIGWIG:
- Rename your final file (substitute YOUR_CELL_LINE and CHROMOSOME):
- Find three of your colleagues with the same chromosome as you. Exchange your BIGWIG files and upload them to the server. Thus you should collect at least 4 BIGWIG files with recognisable names (cell line and chromosome should be present in the file names).
mv <file.genomecov.bw> <YOUR_CELL_LINE-CHROMOSOME.bw>bedGraphToBigWig <file.genomecov.bedgraph> ./GENOME/chrom_sizes.tsv <file.genomecov.bw>Task 6 (0.5 point): Send the screenshot of your directory (as listed by ls command) with collected files.
Expected result of the seminar
- Send the report in canvas in pdf format.
- Answers for all tasks (1-8) are required for the best mark.
- You can get 5 points max for this hometask.
- Commands description, extended (but correct) results, parameters change can help you get extra points. Still, your mark cannot exceed 5 points.
Epigenetics Practice 1 2020
By agalicina
Epigenetics Practice 1 2020
Welcome to Omics Data Analysis Practice on Epigenetics! Today we have our first lecture and seminar. We will cover the general problem of NGS and epigenetics, learn about quality control, ChIP-Seq, ATAC-Seq, DNase-Seq and BS-Seq basics. We'll also learn how to map DNase data and learn a little about epigenetics data association.




