Studying Paramecium's epigenetics with PacBio sequencing


DELEVOYE Guillaume - M2 bioinformatics
Supervisors: Eric Meyer, Mathieu Bahin, Auguste Genovesio
A 340 years old story
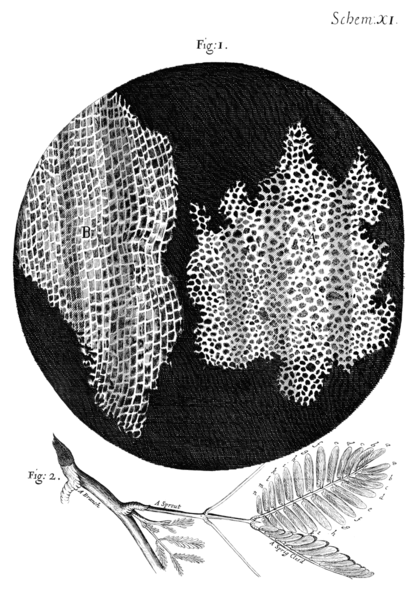

Christopher Hooke
1665 - "Micrographia"
30x microscope
First Cell description




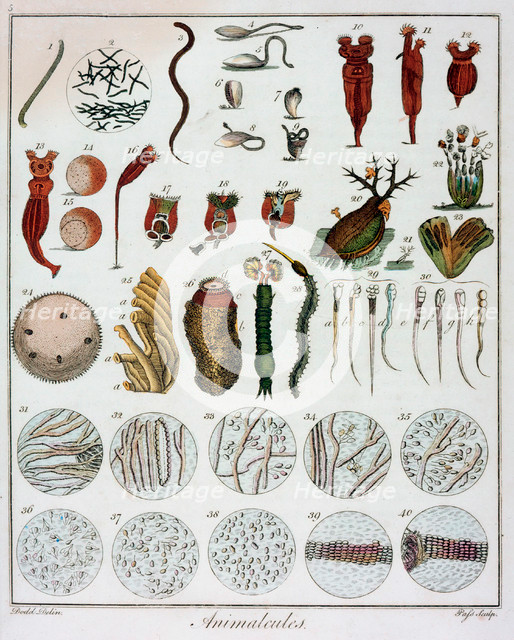
Antoni Van Leeuwenhoek
1678 - "Animalcules"
First observation of single-cell organisms
Infusoria, Paramecium
CIliates: Main discoveries

1862 - Pasteur: Refutation of the spontaneous generation theory with infusoria


1937 - Sonneborn: Non-mendelian inheritance of sexual type in paramecium


Elizabeth blackburn & Carol Greider: 1985 - Telomeres and telomerases on Tetrahymena (Nobel Prize 2009)

Eric Meyer, Sandra Duharcourt
(IBENS, I. Jacques Monod 2014)
Sexual type in paramecium are transmitted by maternal RNAs, not by DNA
How do we see paramecium in 2018 ?
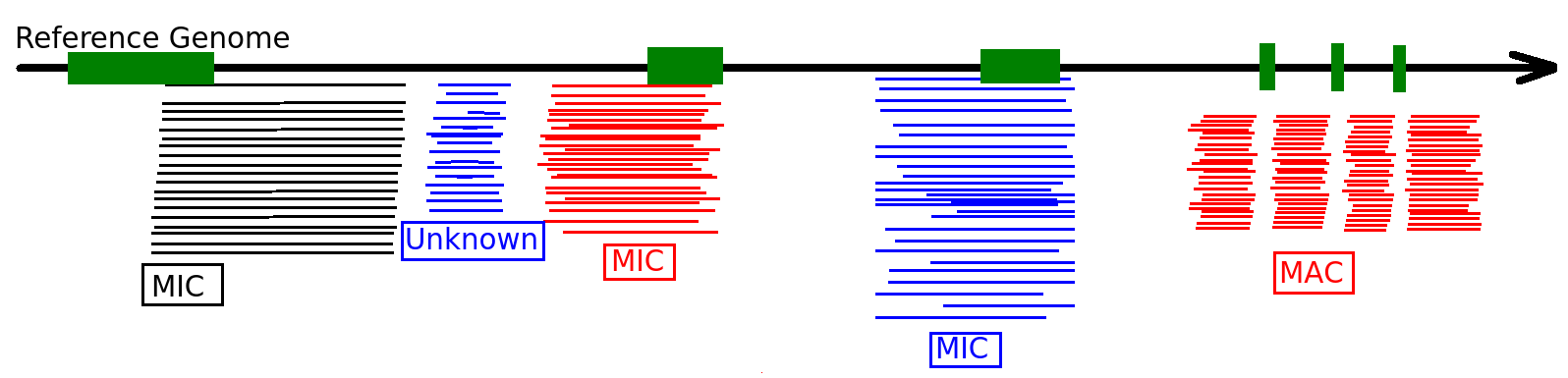
- Eucaryote, Ciliate
- 3 nuclei:
- 2xMIC nuclei (2n)
- Reproduction
- 1xMAC nucleus (up to 800n)
- Transcription
- Partial MIC
- 2xMIC nuclei (2n)
Paramecium's history





3 important events
- Whole genome copy (x3)
- MIC/MAC Differenciation
- PiggyMac Transposase domestication
Survival
MIC/MAC Differenciation
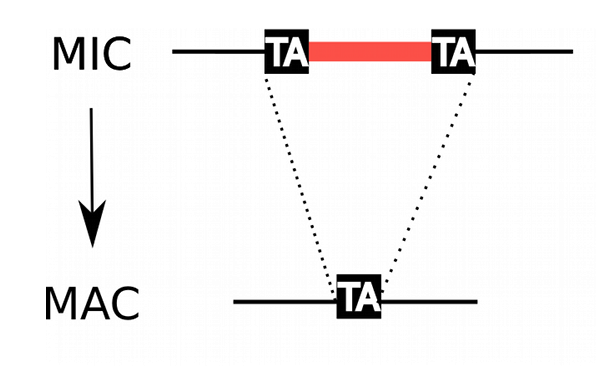
Transposable elements are suppressed in the MAC, where the transcription occurs
Avoids the negative effect of TE
PiggyMac
In 2018, question is...
...How does the cell know where to cut ?
And even more than that
-
WGD potentially implies a lot of ohnologous TE
-
How would the cell still recognize them over the time ?
Genetic drift...
Answer:
It's not fully understood
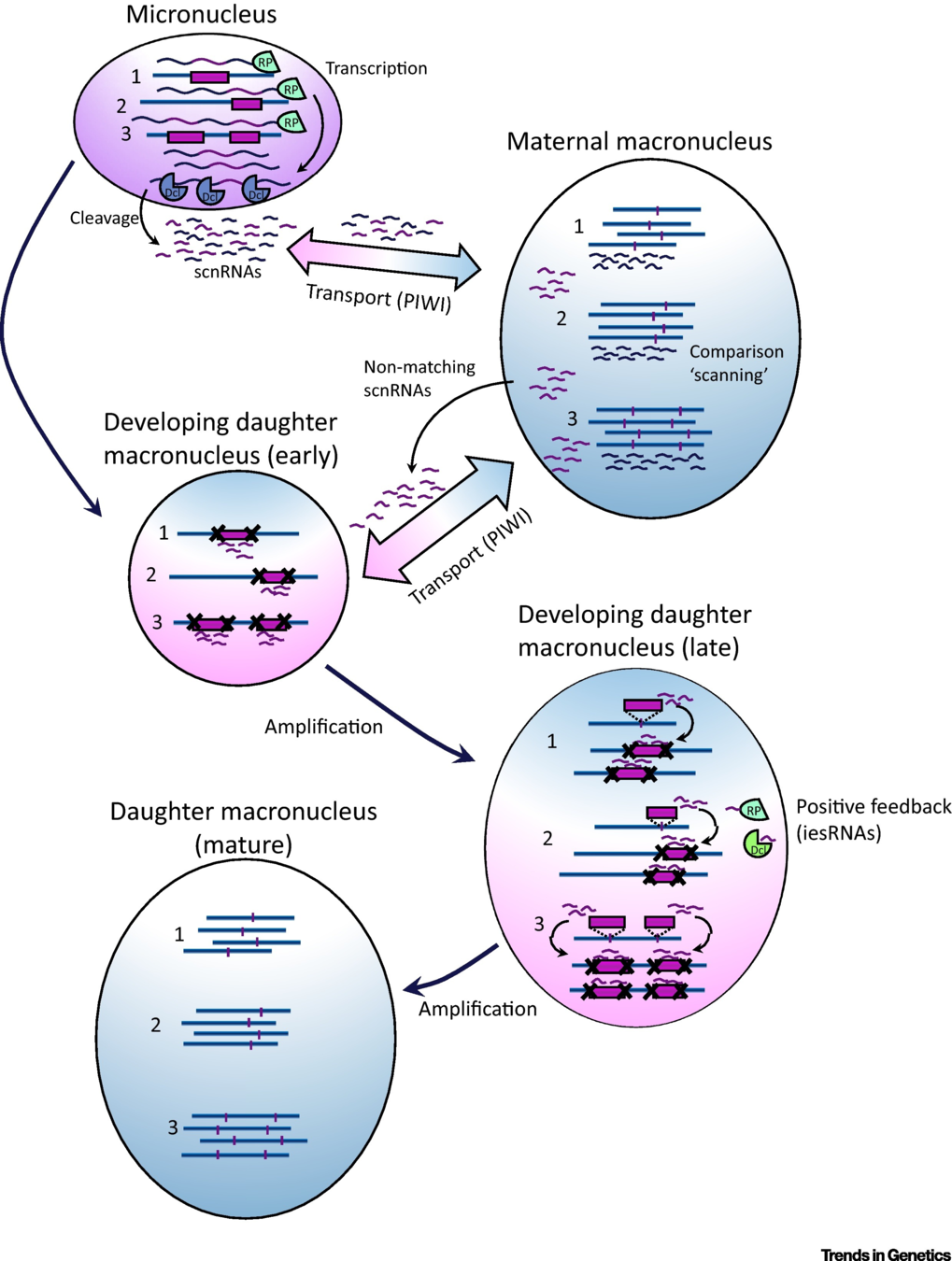
Maternal inheritance
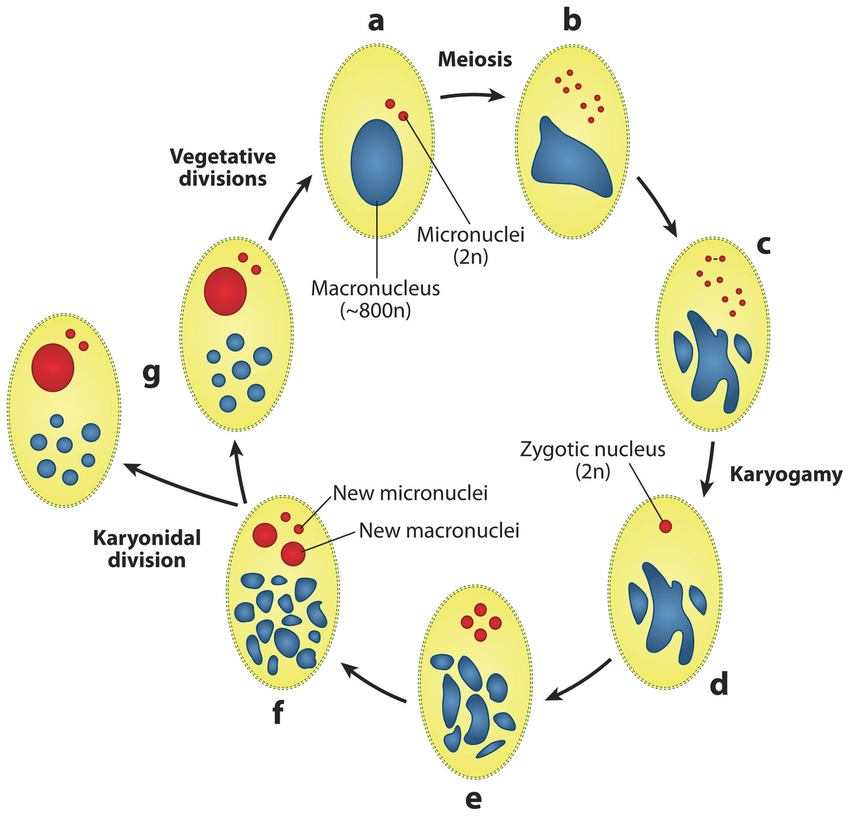
The old MAC (maternal) drives everything during the formation of the new one:
- Active transcription
- Sexual determination (RNA)
- Excision of IES
= Non-mendelian / Cytoplasmic heridity
What we already know about excision
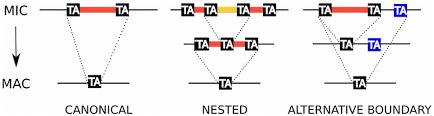
~99% TA-boundary
~ 100% PGM-dependant
"Domesticated enzyme to fight transposons"
Clean cut&Paste mechanism
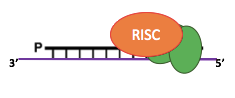
60% SCAN-RNA Pathway
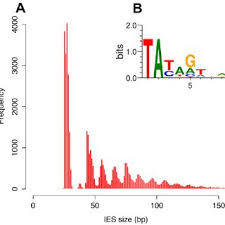
IES excision (3/3): The scanRNA pathway
- SCAN-RNA --> Excision of 60% of IES max (shown by DICER-like silencing)
- Piwi shuttle
- What about the 40% remaining ?
DNA Methylation ?
So, recently in Eric's lab...
Modified bases play an important role in:
- Procaryote's DNA/RNA
- Eucaryote's RNA/DNA
Lots of "orphan-MTases"
Remains highly misunderstood
Seeking 6-mA recognition domain
Protein identification
RNA silencing

Death
--> Role in IES excision ?
Currently being studied
Hypothesis
Please cut me !
Don't cut me !
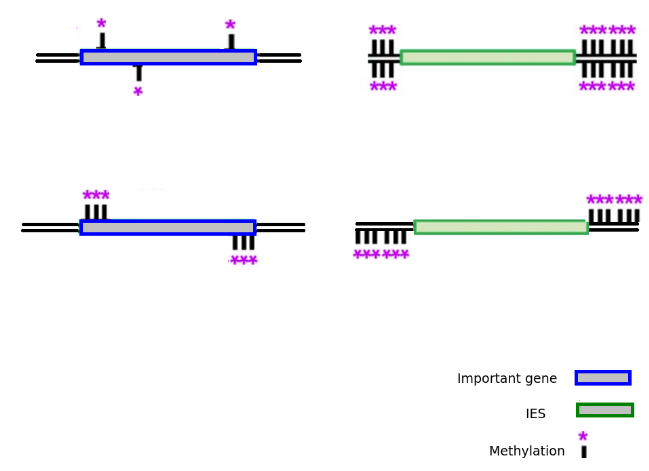
Modified bases: A large landscape of possibilities
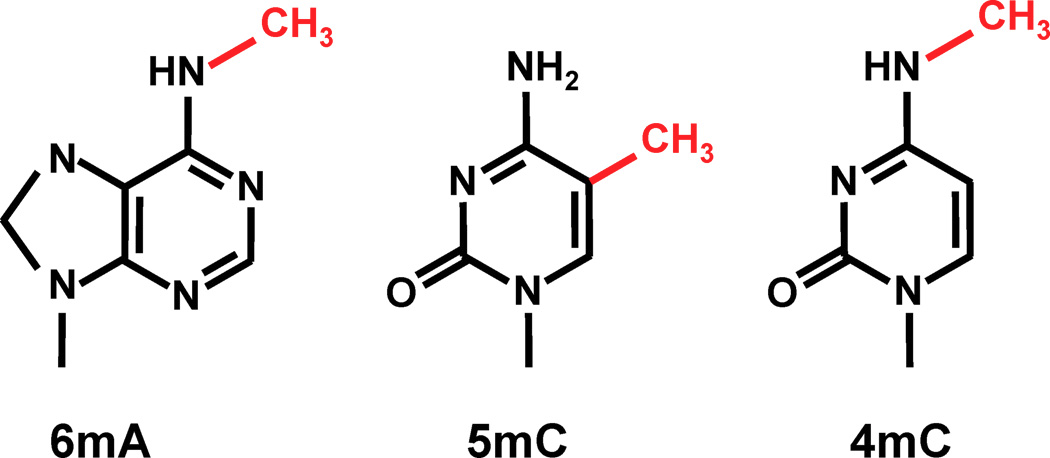
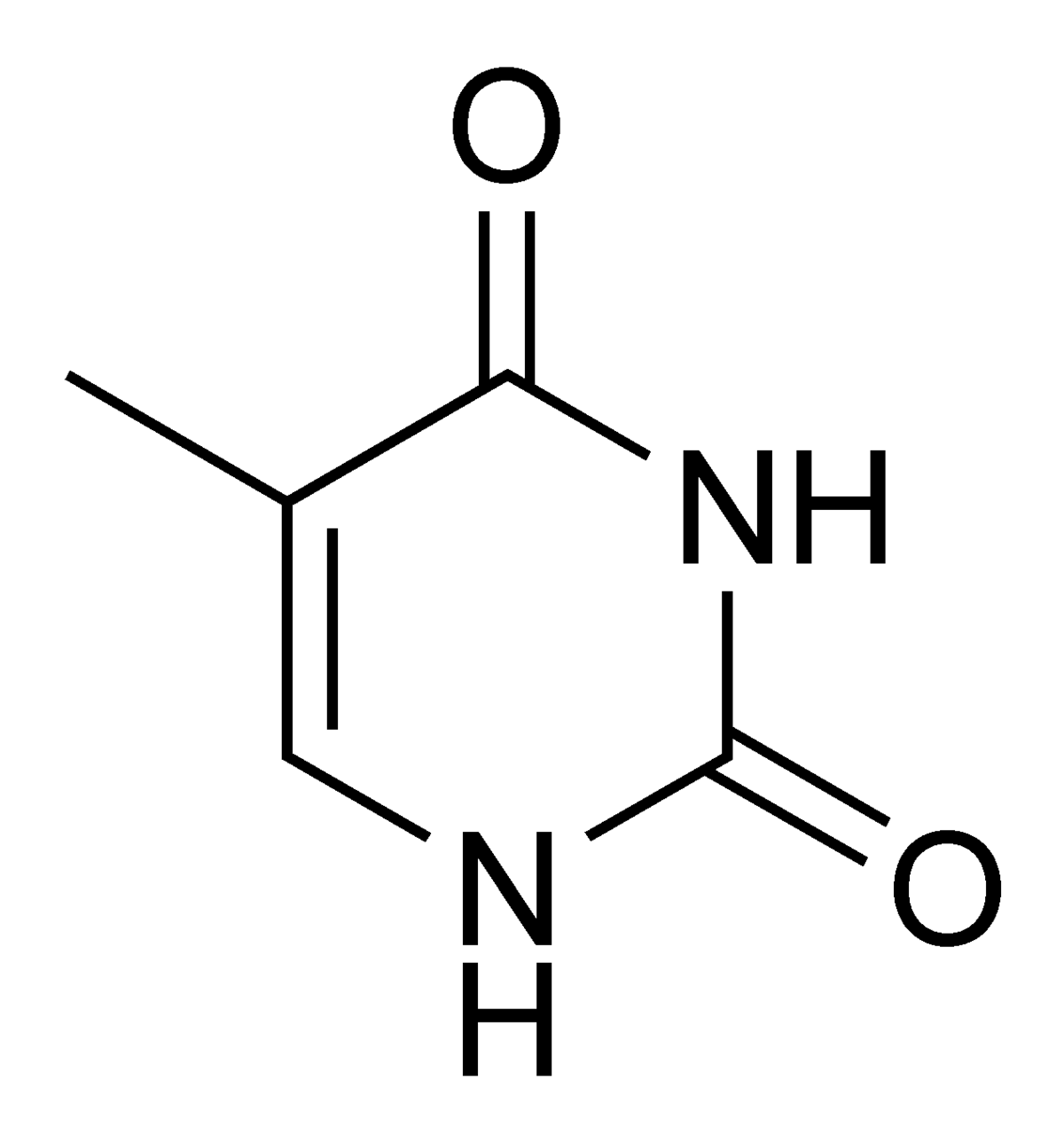
Thymine = 5-methyl-uracile ?
And many others in RNA...
Pseudouridine (ψ) Dihydrouridine (D) m1A m2A m6A m62A i6A t6A Am Ar(p) DHT m3U mo5U s2U s4U iG m1G m2G m22G Gm k2C iC ψC ψiC m3C m4C m5C m42C Cm ac4C s2C m1I Im...
<-- 3 known most frequent
methylations in DNA
Challenges
-
Sequencing : Sorting MIC and MAC
-
Not experimentally possible
- Or we loose the methylation information...
-
Not experimentally possible
-
98% of IES are < 200 pb: "Long read" needed (to sort)
- MIC: 1/400 to 1/800 sequences only !
- Resolution needs:
- Single molecule
- Single strand
- Single nucleotide
- Identification of modified bases, including unknown ones
- 3 Whole genome duplication ! (mapping)
PacBio sequencing meets all expectations
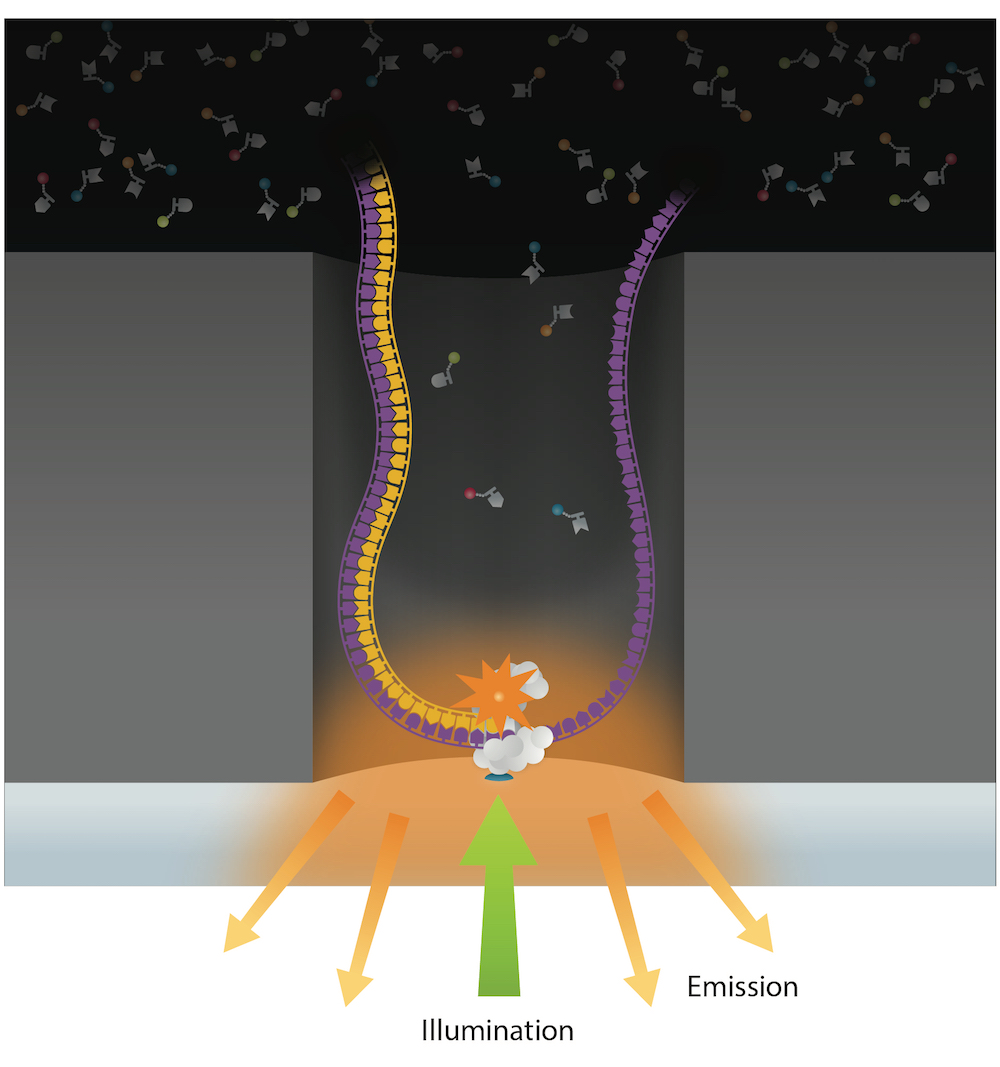
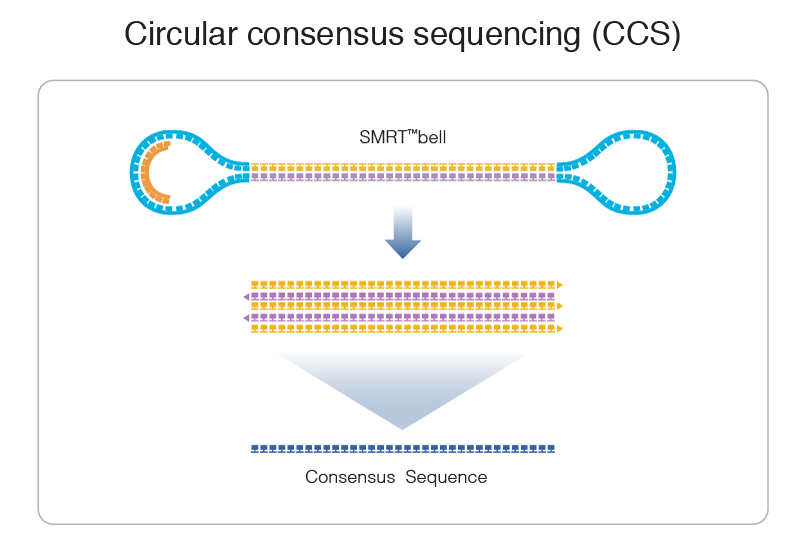
99% accuracy
Max
75% accuracy
Methylation analysis needs local > 25X
Trained model (ML) allows detection of suspect downturns of polymerase (function of the -3/+8 nt context) --> IPD are captured
SMRTLink
A very user-friendly browser interface
Advanced analysis are impossible
--> Command-line tools required
Theory: Experimental strategy
Classical PacBio Approach: Higher depth by overlapping the holes
Our approach: Shorter, real single hole analysis, much more passes
We ~always have either 0X or >>> 25X
But reality can be cruel...

Murphy's Law statistically hits a lot if you're trying 500.000 times

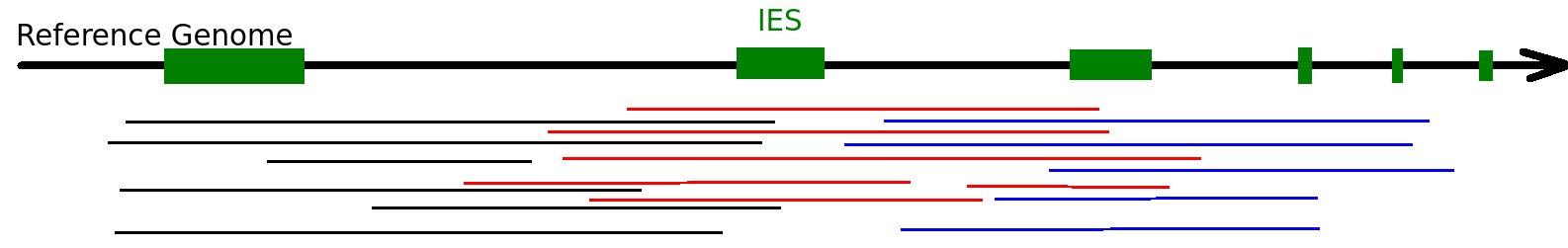
Other cases:
- Some IES are excised, some others not
- Partial excision...
- Multiple mapping that are ~ equivalent
- Not the same mapping position between MAC and MIC
- etc...
Step 1: Sorting the sequences
Create and align the consensus
0 - Quality filter (Z-score)
1 - Create the consensus (=CCS)
2 - Map the CCS on MAC / MAC+IES (BLASR)
--> Only the best alignment reported (forced)
3 - Filter 90% identity for either MAC or MAC+IES
4 - Compare the mapping
Reminder: CCS are expected to be somewhat around 99% accuracy
Differential mapping
Sequences that come from MIC
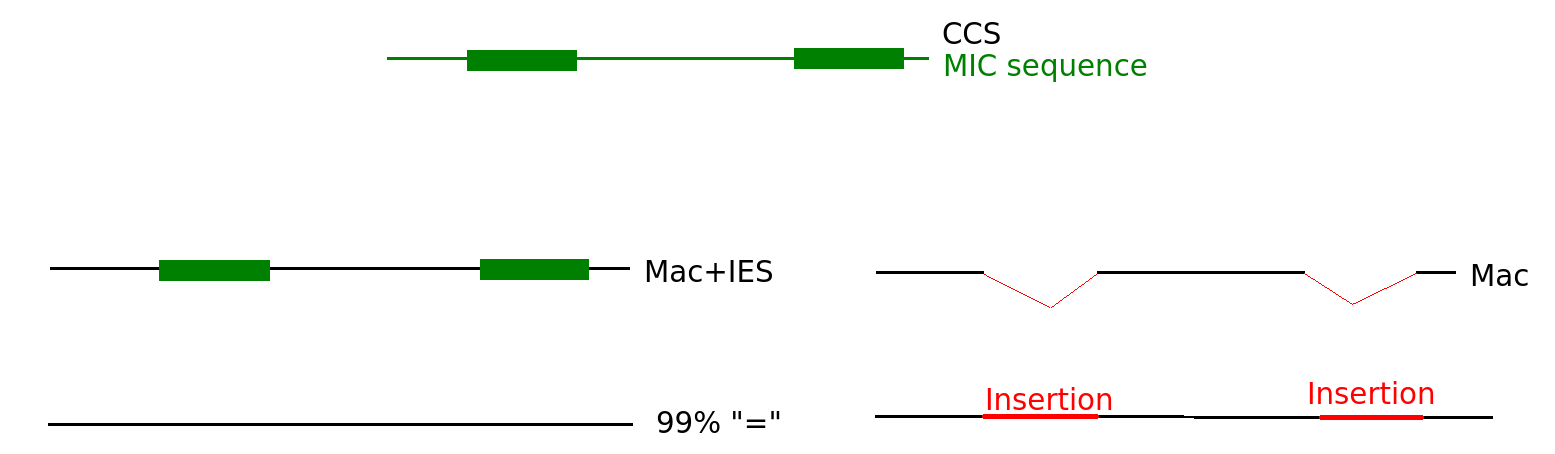
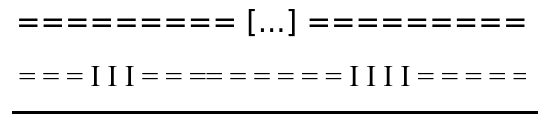
N changes
"=" becomes "I"
Threshold N = 27
At least N = 27 positions map on IES
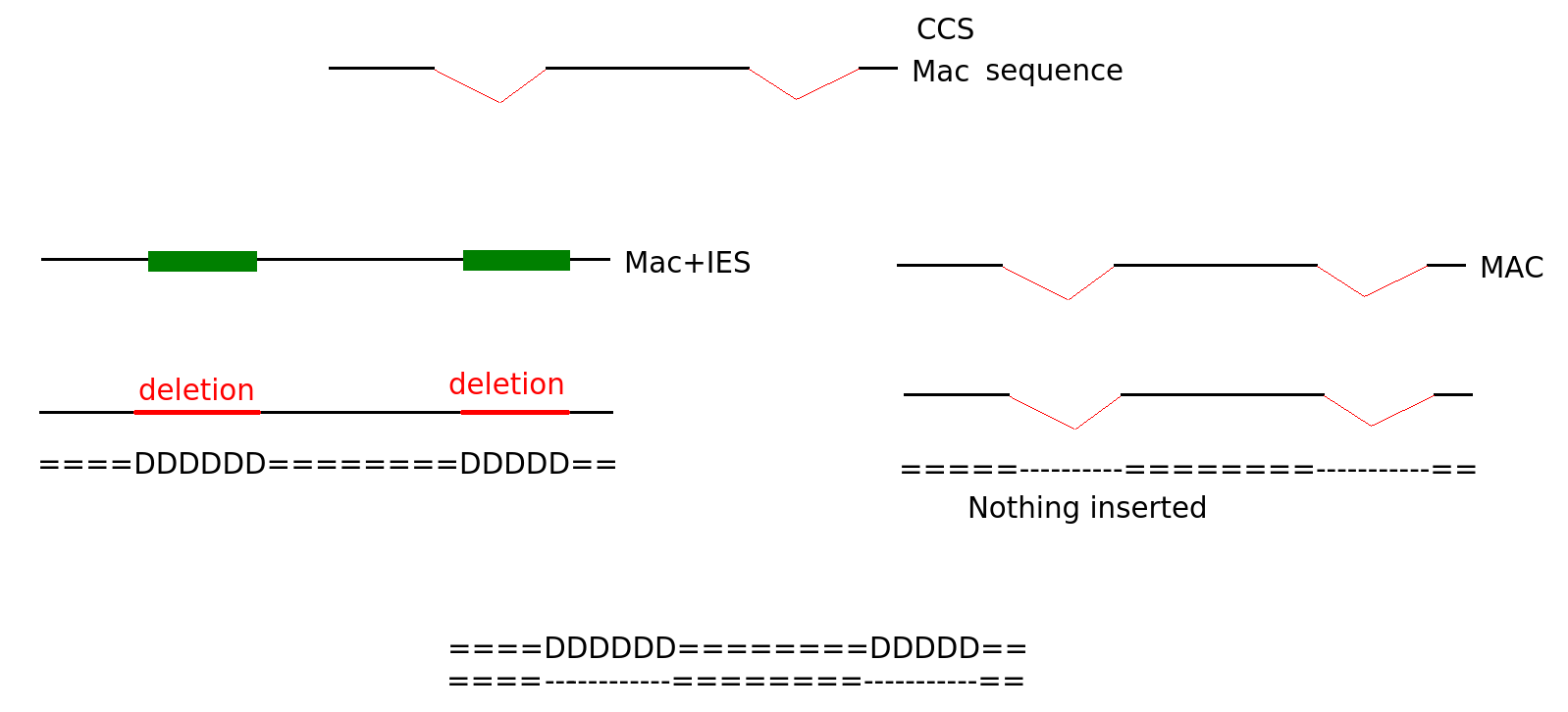
Diffential mapping
Sequences that come from MAC
A least 2 positions have to be on a junction
N > 27 changes
"D doesn't match I on expected IES"
Other cases:
-
Put in "unknown" group:
- Low identity CCS
- N number of changes under threshold (27)
- CCS mapping on totally different scaffolds, with about the same scores/identity
- CCS that didn't map on any IES or IES junction
Everything else:
"Trash"
Visual inspection
- First intuitive checking
- Stringent --> Better so ! (FP are avoided)

Rigorous Benchmarking
-
For now mostly visual checking, but:
- Automatic checking of presence/absence of IES/junction
- Coherent number of sorted/unsorted DNA
- Soon a rigorous benchmarking (TP/FP/TN/FN): PBSIM

Step 2
Recreating the corresponding .bam files
- Can need a lot of RAM for the hashed lookup tables (python dicts)
- Ok on the cluster with higher RAM usage
-
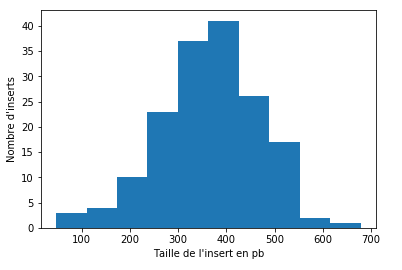
218 holes of MIC inserts that cross an IES
- All greatly covered (>25X to 100X)
Step 3:
Single hole Analysis of methylation
Remember
Everyone else's PacBio usage:
Eric, a weird scientist:
Pauses of the polymerase:
A statistical problem
Sometimes the polymerase is "Lazy" and stops for a VERY long time --> Really annoying (methylation analysis)
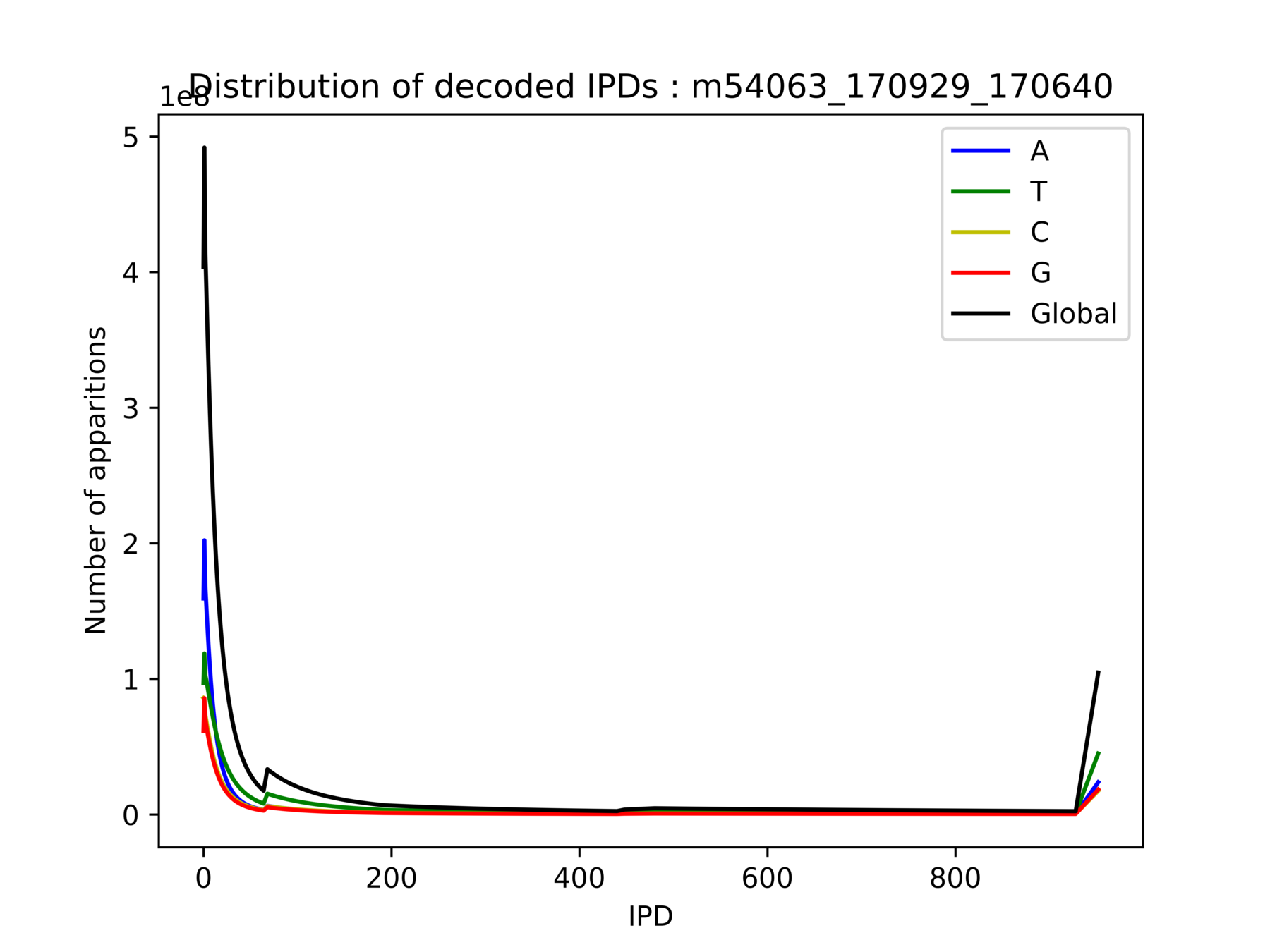
Outliers:
- Very big compared to others
- Really disturb all computations related to the mean:
Modification detection, modification identification, p-value, PHRED-score...
Outliers: How to deal with them ?
- We want to eliminate very big values
- We don't want to loose high IPDs correlated with methylation
- Trade-off
What's in PacBio's statistical blackbox

How PacBio handles it
--->> Values are "capped":
cappingValue = max(99th chunk, 4* modele, 75th local percentile)
- We need the mean
- p-value, PHRED... Rely on the mean
- We cannot take the median
- Some context of nucleotides are naturally slower than others
- Outliers are relative !!
- Context is important
- They should represent far less than 1% of all IPDs values
For every position in the reference:
What if we analyze hole/hole ?
cappingValue = max(99th chunk, 4* modele, 75th local percentile)
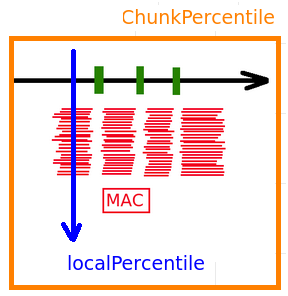
Probably it doesn't change much
But it's rigorously not the same
Someone already worked on it over the last months


Unfortunately
- Old .h5 format
- No p-value, no PHRED-score, no identification
- Many statistical refinments, but not those we need
The solutions
- Median <-> Maybe not ideal ?
- Let's pretend we're doing it normally
- Pre-compute all the capping values
- Re-use those capping values in a true single-hole analysis
>>> This is also how the recently publication of Beaulaurier works (with hd5)
Hacking PacBio
PacBio "kineticsTools" could work only:
- On whole genome
- Without giving all the values it computed
Now it's properly hacked:
- Spits every capping values so that we can study them
- Works on little chunks / Single hole (much faster)
- Properly hacked to work in Parallel (Multiprocessing)
- Not Condor ready
- Up to 80 processors on Bioclusts02
- ~ 10s/hole on Bioclusts02
A first try
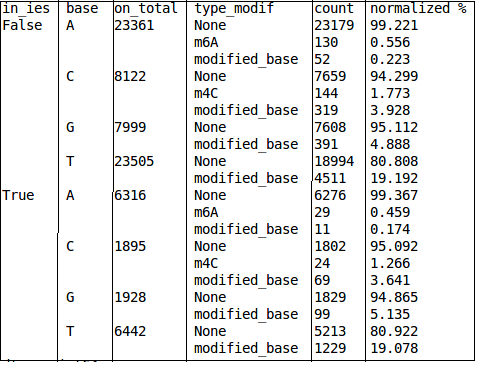
Strand issue (BLASR) to fix: Only strand 0 is available
Quality threshold
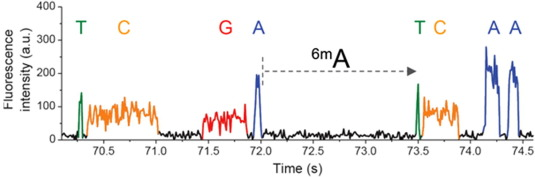
Real results
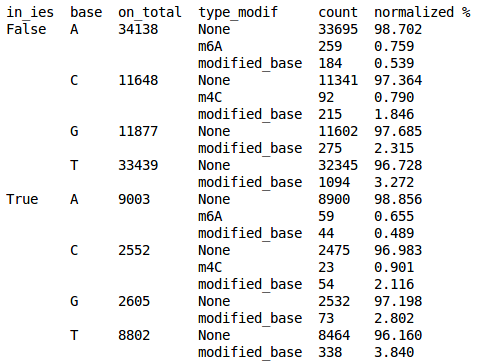
What remains to do
- Annoying behaviours of BLASR to fix
- BLASR doesn't perform great for "one position only"
- Alternatives exist... I didn't investigate it yet
- Benchmark the sorting
- Compare modified-capping VS naive approach
- Motif analysis
- Stats SCAN-RNA vs independant ++
- Other samples
- Nextflow for reproductibility
Studying Paramecium's methylation with PacBio sequencing
By biocompibens
Studying Paramecium's methylation with PacBio sequencing
Lab meeting - 19/06/18



