Single-molecule DNA methylation analysis by SMRT sequencing of short inserts
DELEVOYE Guillaume - 1rst year PhD student
Supervisors: Eric Meyer, Mathieu Bahin
ANR Meeting 08/04/19
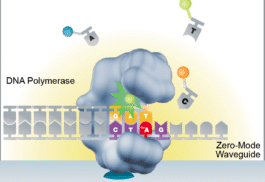
Modified bases: A large landscape of possibilities
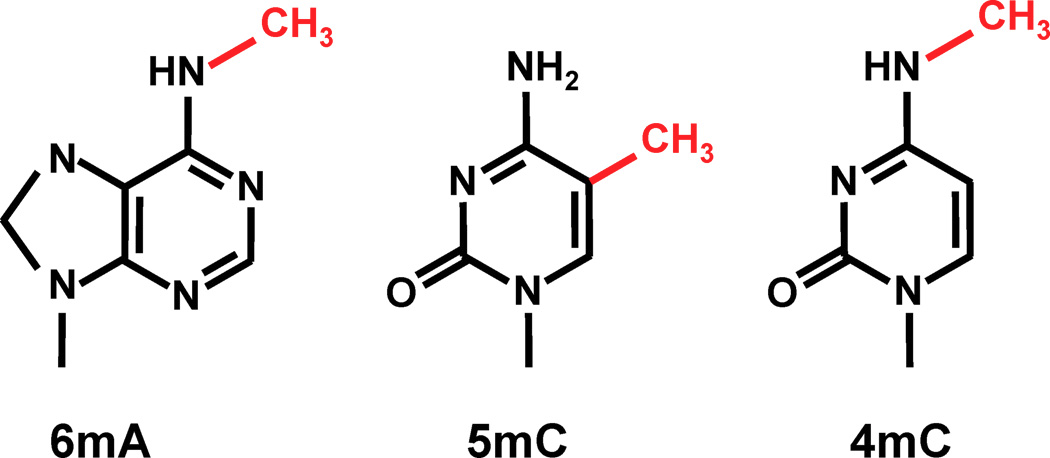
Thymine = 5-methyl-uracile ?
<-- 3 known frequent
methylations in DNA that we can detect with PacBio
25X
25X
250X
PacBio sequencing
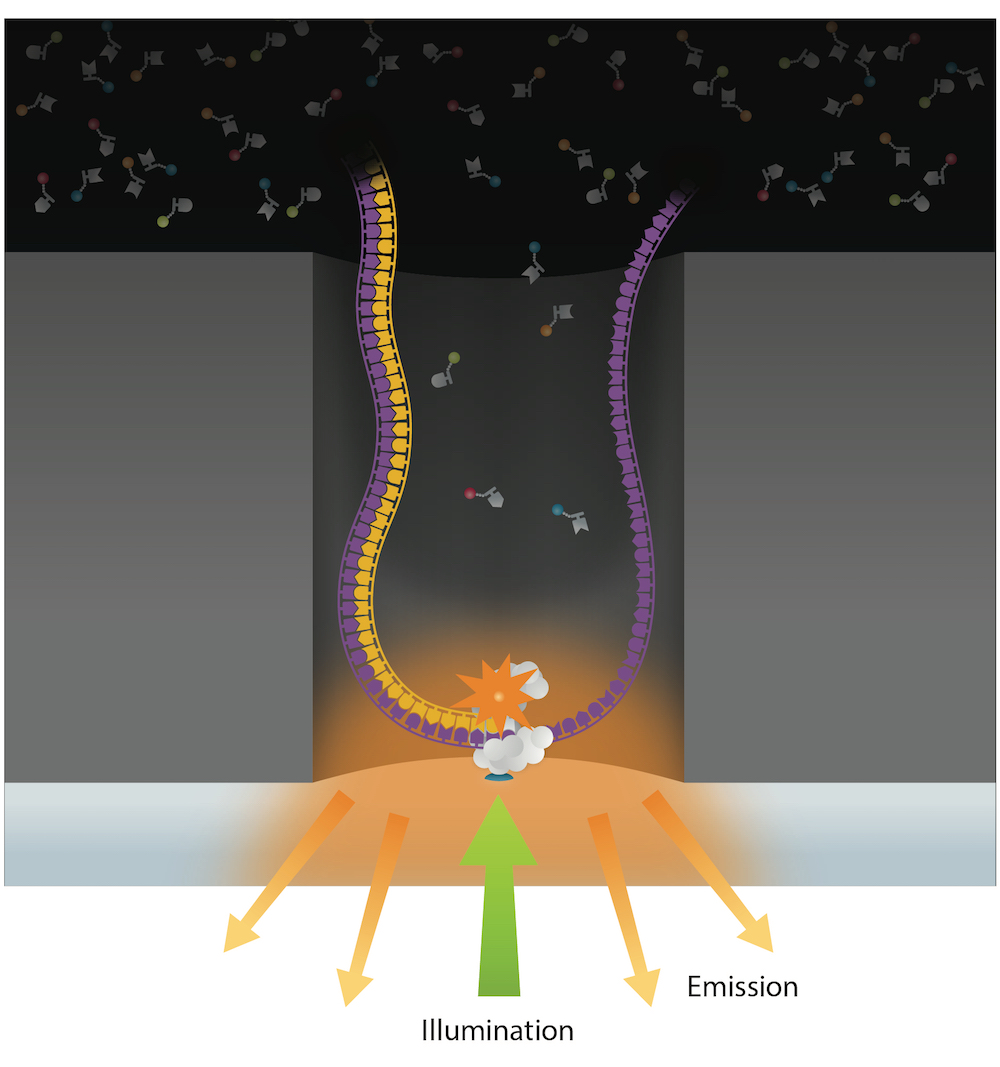
99% accuracy
Max
75% accuracy
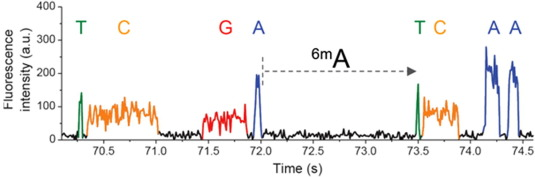
Unusual Slowing: Modification Score (Qv)
Kinetic signature: Identification score (IdQv)
Trained model (ML) allows detection of suspect downturns of polymerase (function of the -3/+8 nt context) --> IPD are captured
~ PHRED scores
Theory: Experimental strategy
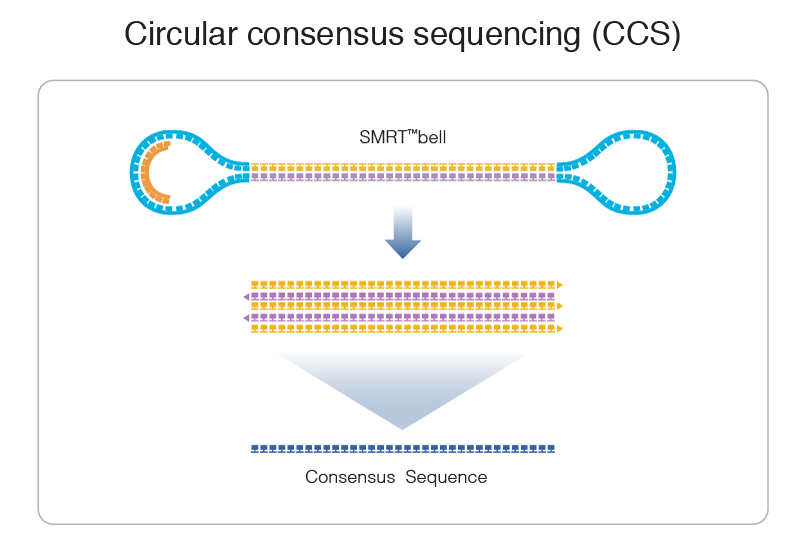
Classical PacBio Approach: Higher coverage by overlapping the holes
Our approach: Shorter, real single hole analysis, much more passes
We ~always have either 0X or >>> 25X
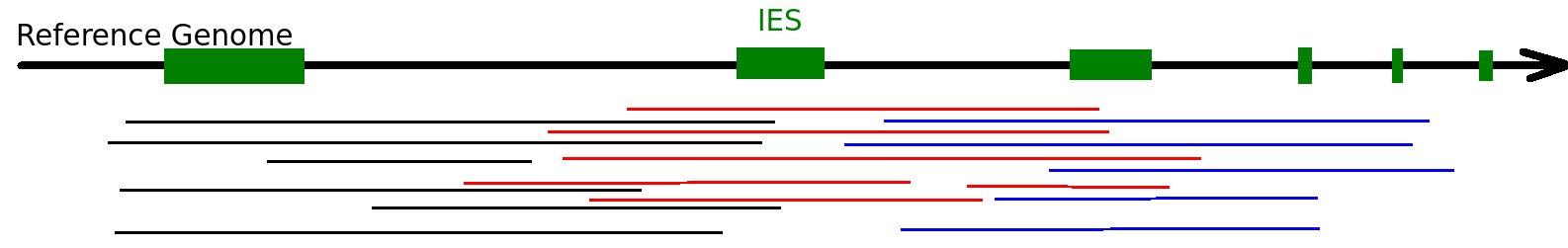
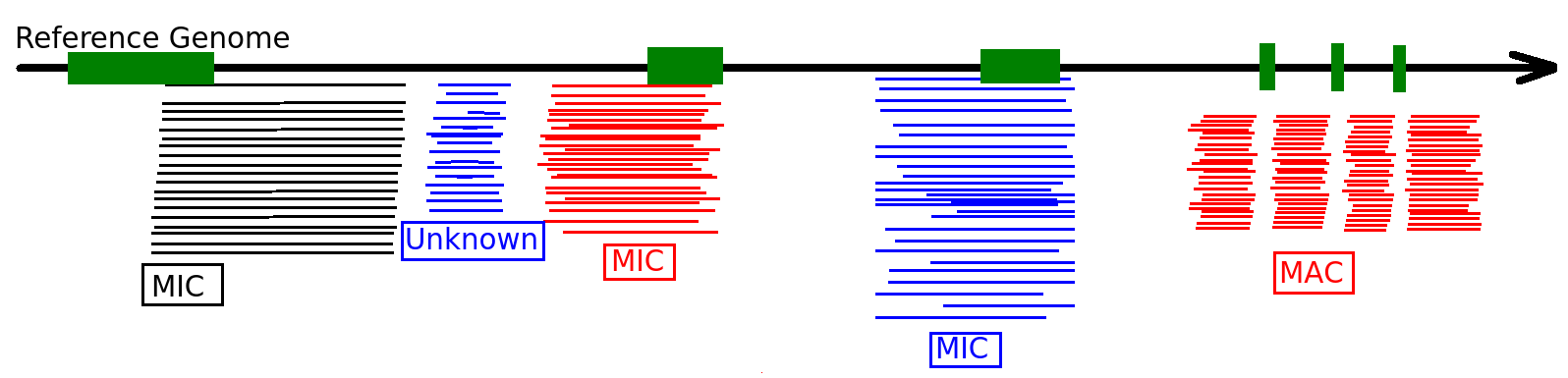
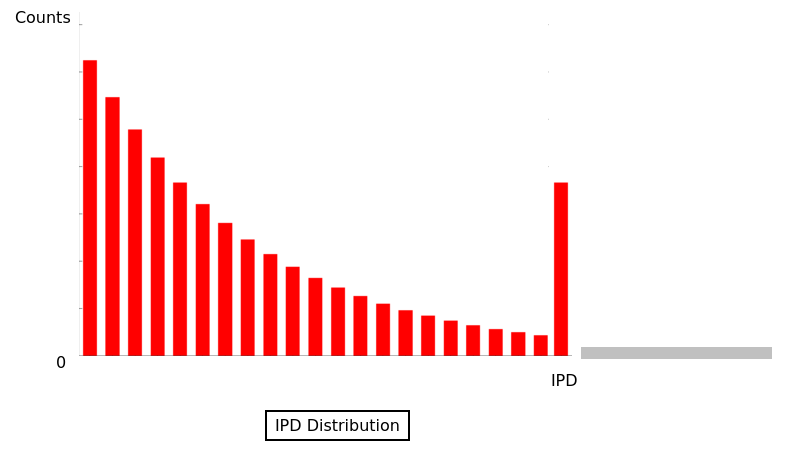
Majority of MDS come from the MAC
Step 1: Sorting the sequences
Some technical issues occured on the way

Murphy's Law statistically hits a lot if you're trying 500.000 times
Create and align the consensus
0 - Quality filter (Z-score)
1 - Create the consensus (=CCS)
2 - Map the CCS on MAC / MIC / MAC+IES (BLASR)
--> Only the best alignment reported (forced)
3 - Filter at >99% identity on at least one genome
4 - Compare the mapping
Reminder: CCS are expected to be somewhat around 99% accuracy
Differential mapping
Sequences that come from MIC
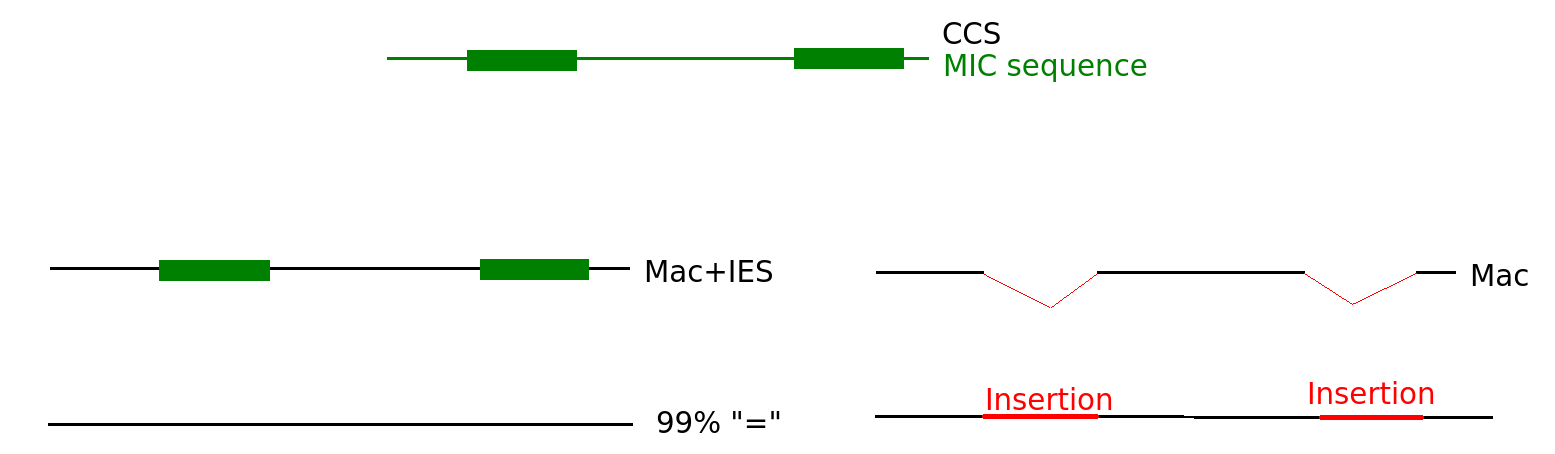
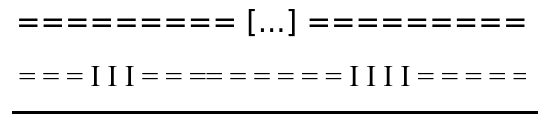
N changes
"=" becomes "I"
Diffential mapping
Sequences that come from MAC
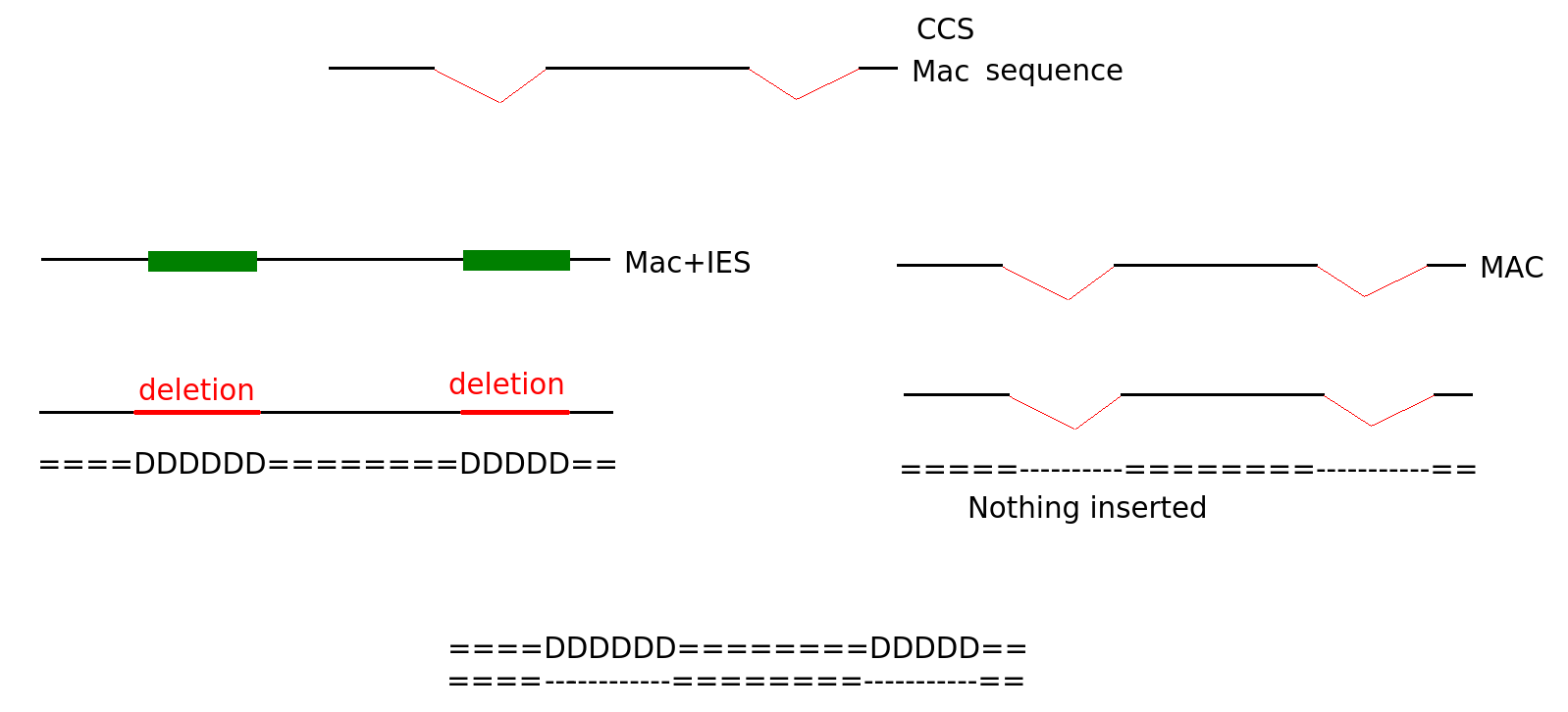
Step 2: Removing the pauses
A lazy polymerase
The polymerase makes random pauses, that are not linked to DNA modification
What's in PacBio's statistical blackbox

How PacBio handles it
--->> Values are "capped":
cappingValue = max(99th chunk, 4* modele, 75th local percentile)
- We need the mean
- p-value, PHRED... Rely on the mean
- Very sensitive to outliers
- Pauses must be removed !
- Some context of nucleotides are naturally slower than others
- Context is important
- Pauses should represent far less than 1% of all IPDs values
For every position in the reference:
Should the capping be the same when using a single hole approach ?
cappingValue = max(99th chunk, 4* modele, 75th local percentile)
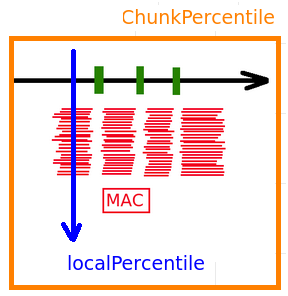
It doesn't change much
But it's rigorously not the same
Step 3
Analyse the methylation
What threshold scores should we use ?
The score threshold
The modification/identification scores are sold by PacBio as PHRED scores
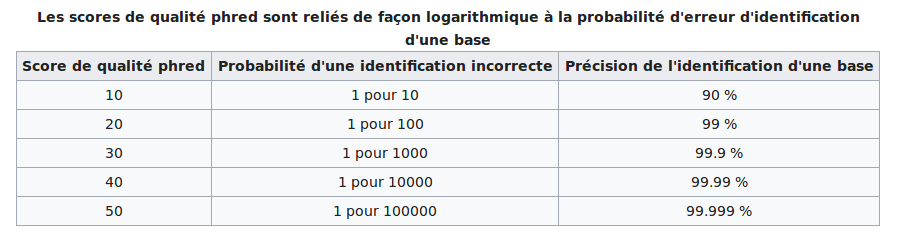
For our experiments: we should have about ~30% of modified bases if this is true
--> Modification scores are NOT prhed scores (at least in our case)
Modification scores overall
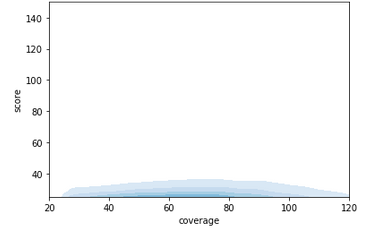
Score distributions: The case of the adenines
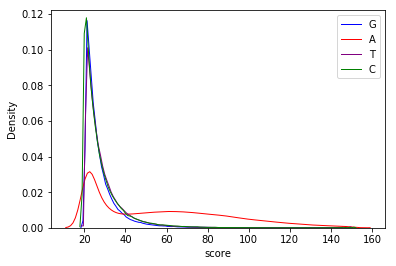
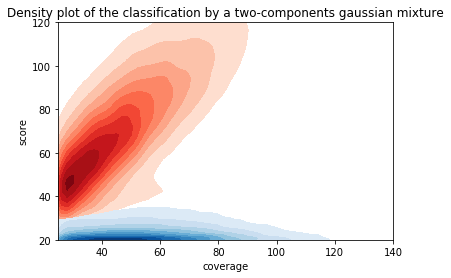
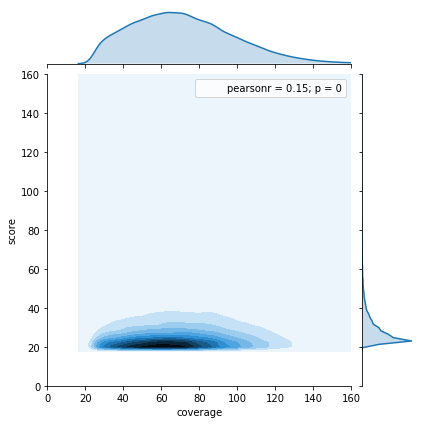
Can we apply the GMM to other samples ?
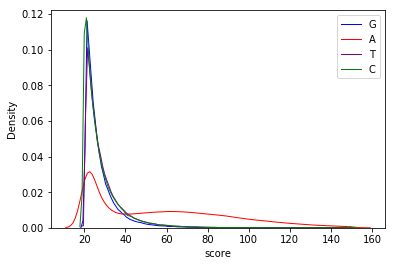
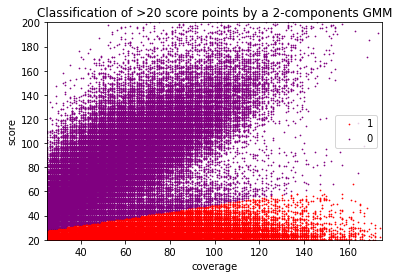
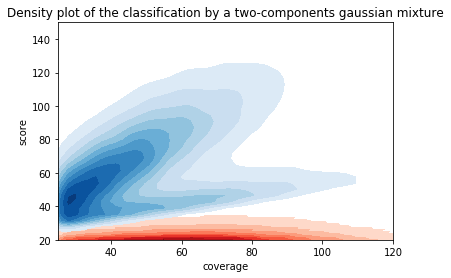
Score distributions: The case of cytosines
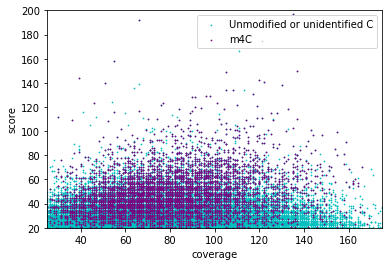
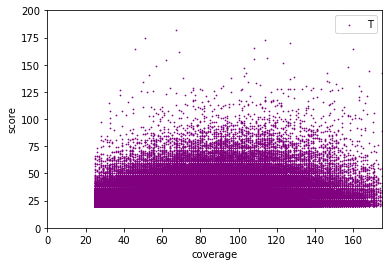
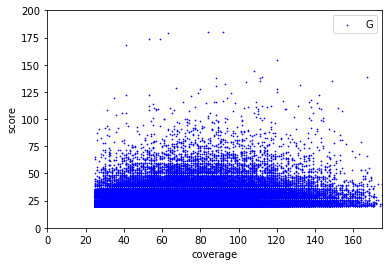
Score distributions: The case of G and T
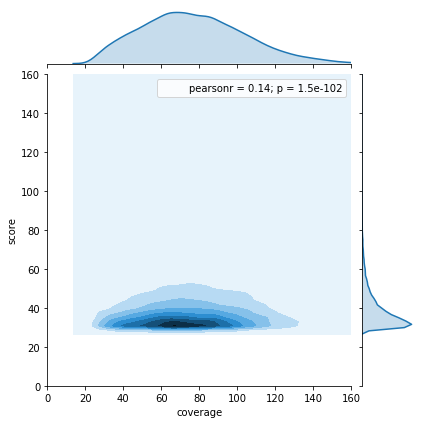
Available Data
HT2
HT6
HTVEG
MAB
MT1A-1B
MT1A-1B-2
MT2
NM9_10
NM4_9_10
WT
Silenced
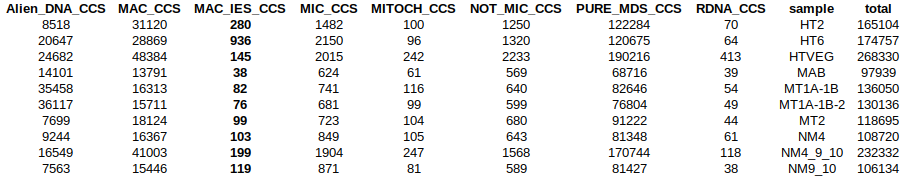
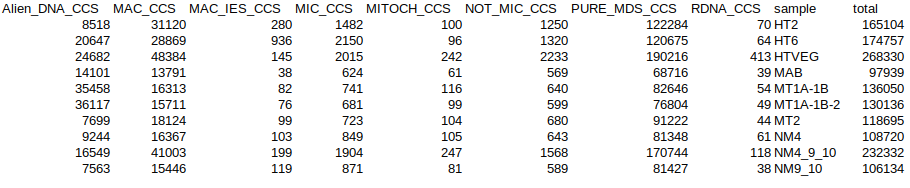
Sorting stats
Using only the GMM
Adenines
All A in GMM
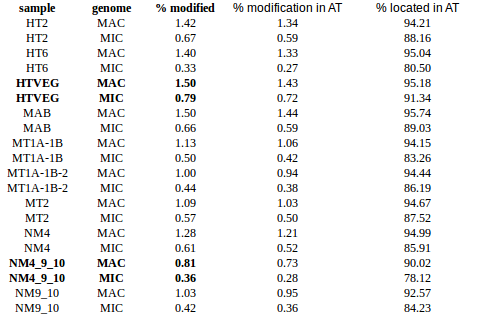
- Same MIC/MAC in AT
- No difference between experiments
- ~95% methylated symetrically
All A in GMM
out the AT
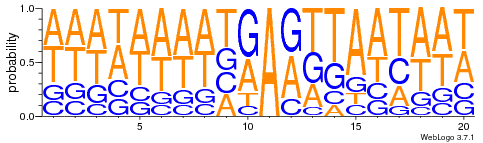
HTVEG
~ Same for every silencing experiment
Lack of sequences (~50 VS ~2.000) don't really allow comparaison between MIC and MAC
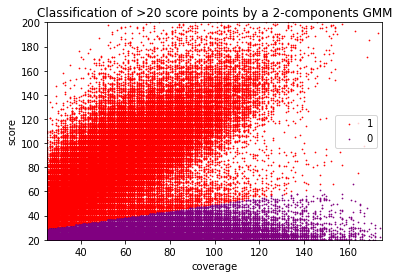
Using idQv20 + GMM
Adenines
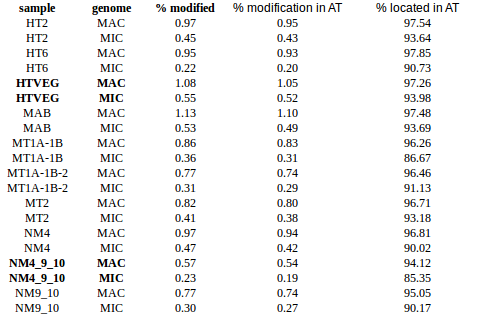
GMM + idQV20
- Raises the % located in AT
- Methylated fraction goes down to 0.9-1%
- NM4-9-10 goes down to 50% in the MAC
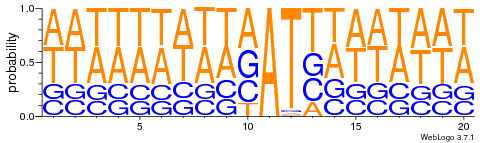
- Logos don't change significantly
Cytosines
Qv20/IdQv20
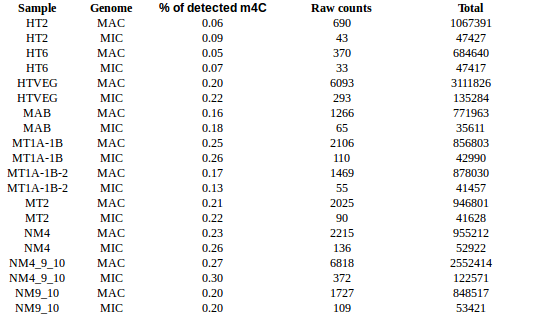

Logo from HTVEG MAC
Identical everywhere
Conclusion
Number of MAC_IES sequences: will it be enough ?
Capping ok
In silico control --> Experimental
Coverage threshold single-hole ?
Threshold is still a +/- open question
Diminution m6A in some silencings, but which one disappears ?
2.5% m6A in MAC <-> Bad calibration ? MDS missing ? Lack of sensibility ?
NM4_9_10 --> 10% ? Recalibrage par optimisation ?
m4C signal: HT2 = HT6 < HTVEG
Sorting stats 2
(ANR) Single-molecule DNA methylation analysis by SMRT sequencing of short inserts
By biocompibens
(ANR) Single-molecule DNA methylation analysis by SMRT sequencing of short inserts
Lab meeting - 19/06/18



