













































Inês Mendes
Bioinformatics PhD student.
Thesis Comittee
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
15th of September, 2020
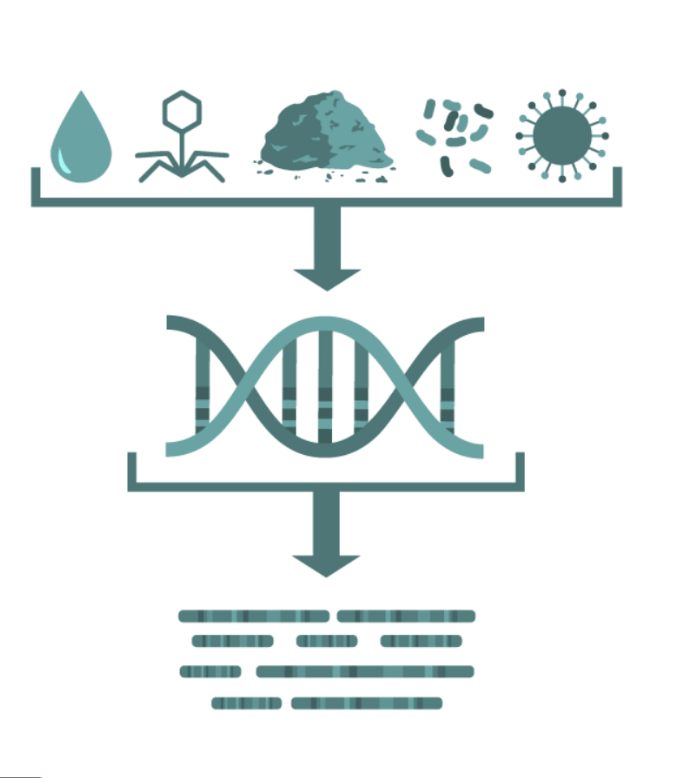
Random "shotgun" sequencing of microbial DNA, without selecting a particular gene.
Promising methodology for obtaining fast results for the identification of pathogens and their virulence and antimicrobial resistance properties without the need for culture.
Who is there? - Taxonomic identification
What are they doing? - Virulome, Resistome, Functional Annotation
Who is doing what? - Functional Assignment
Main Goals
Funding
Promoters & Host Institutions
Precision, Sensibility & Performance
(Culture + Maldi-TOF)
11 Metagenomic Samples - Fluid & Tissue
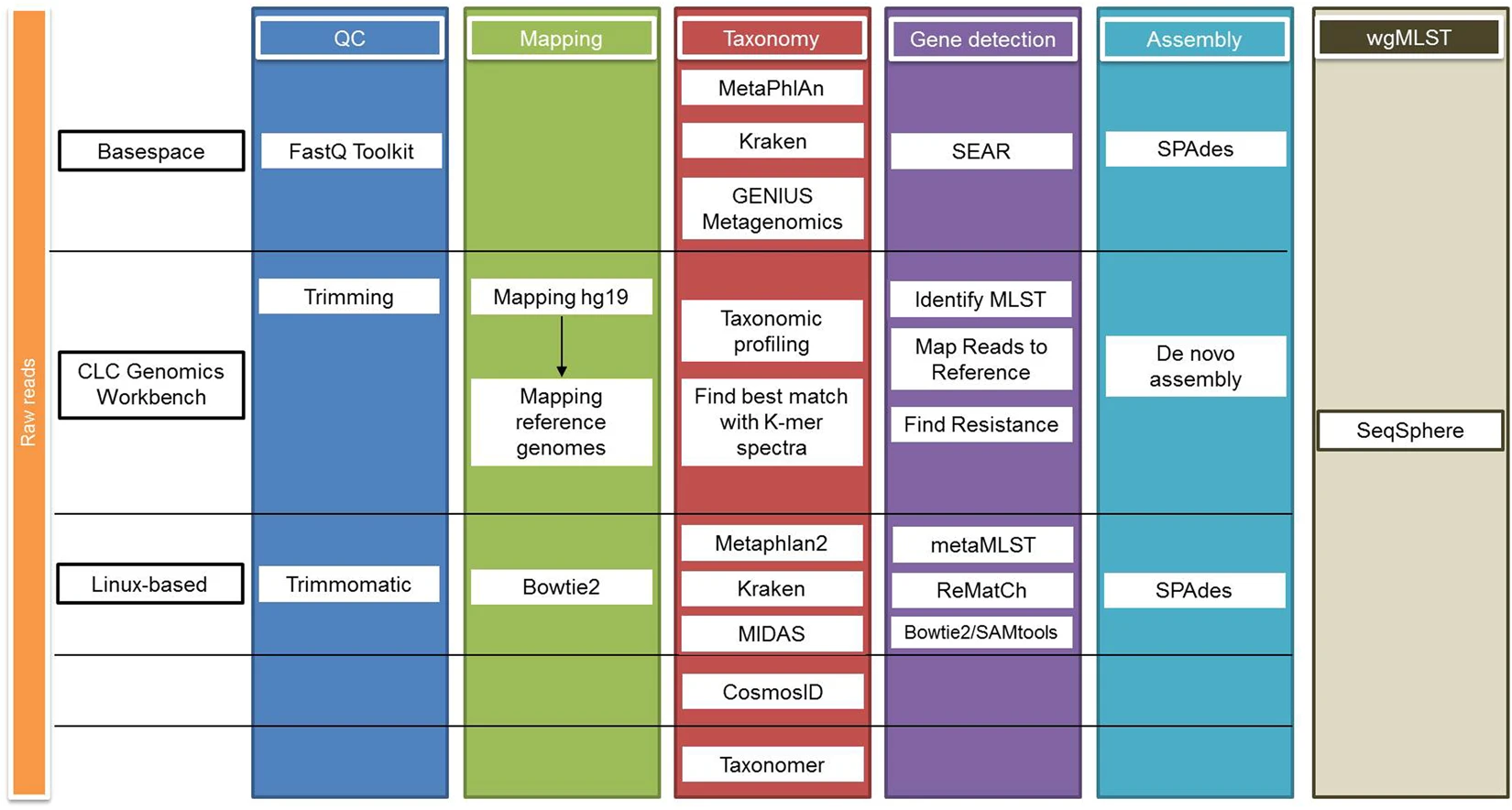
Major issues
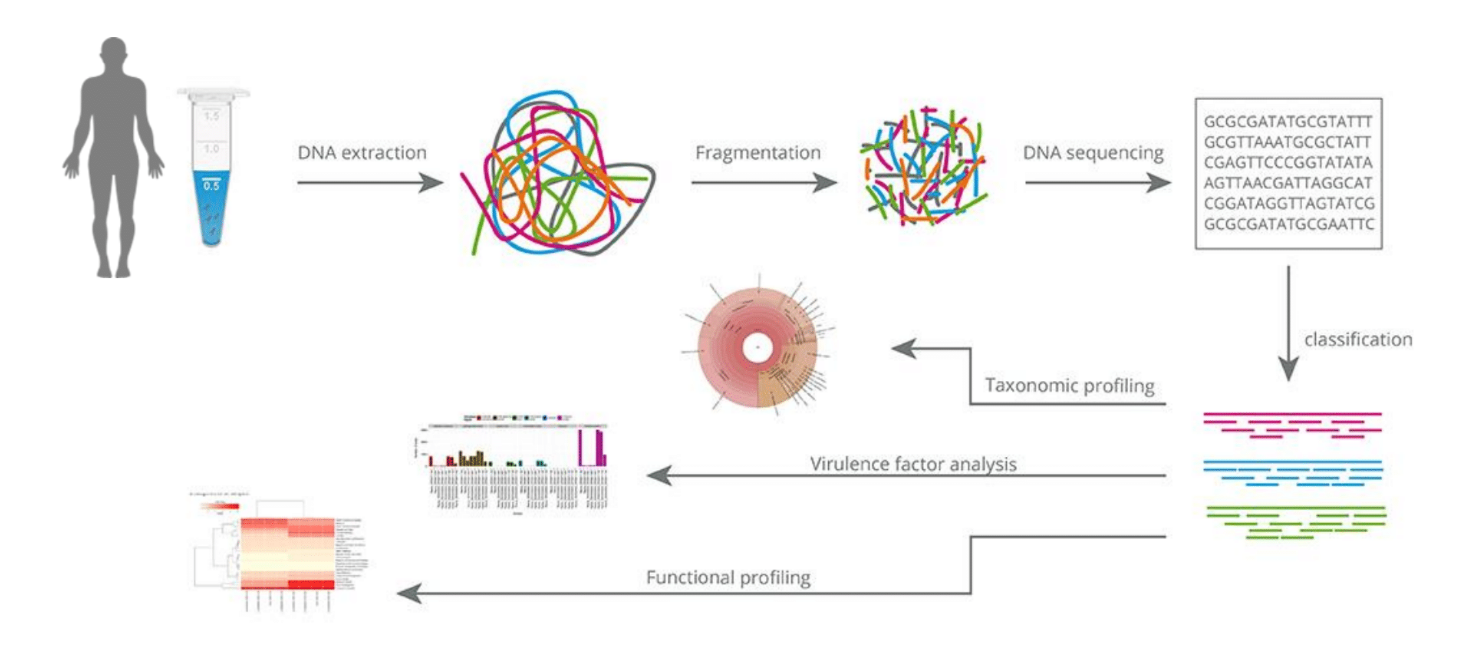
Clinical Shotgun Metagenomic Analysis
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
15th of September, 2020
The needs:
Writing of pipelines in python/perl/shell scripts circa 2000, colorized.
Workflows in the Paleolithic era:
The game changing combination of workflow managers and containers:
Workflows in the Modern era:
Workflow based development
Component based development
Components are modular pieces with some basic rules:
Component A
- Input/Output
- Parameters
- Resources
Component B
- Input/Output
- Parameters
- Resources
With this framework, building workflows becomes simple:
flowcraft build -t 'trimmomatic fastqc spades pilon' -o my_nextflow_pipelineResults in the following workflow DAG (direct acyclic graph)
It's easy to get experimental:
flowcraft build -t 'trimmomatic fastqc skesa pilon' -o my_nextflow_pipelineSwitch spades for skesa
Forks
Connect one component to multiple
Secondary channels
Connect non-adjacent components
Extra inputs
Inject user input data anywhere
Recipes
Curated and pre-assembled pipelines for specific needs
Multiple Raw Input Types
Not limited to paired-end FastQ or Fasta
Dynamic Input in Components
One component, multiple inputs
Expand Building Features
New merge operators
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
15th of September, 2020
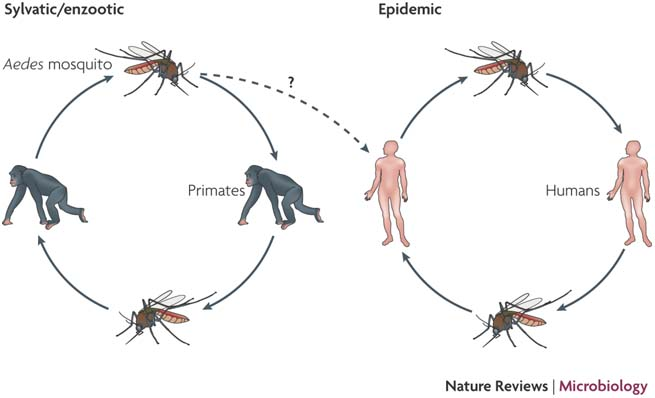
dengue virus genotyping from amplicon and shotgun metagenomic sequencing
doi:10.1038/nrmicro1690
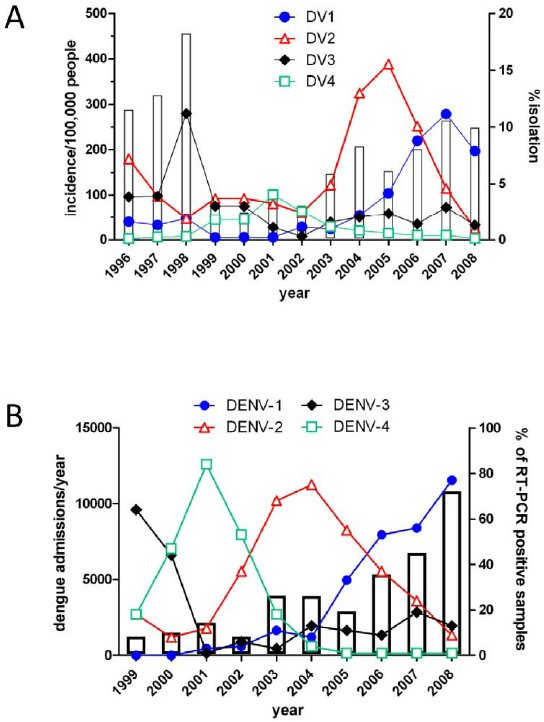
Sequential infection increases the risk of a severe form of the infection - dengue hemorrhagic fever.
Dengue hemorrhagic fever:
https://doi.org/10.1371/journal.pntd.0001876.g002
https://doi.org/10.1371/journal.pntd.0000757
Thailand
Viet Nam
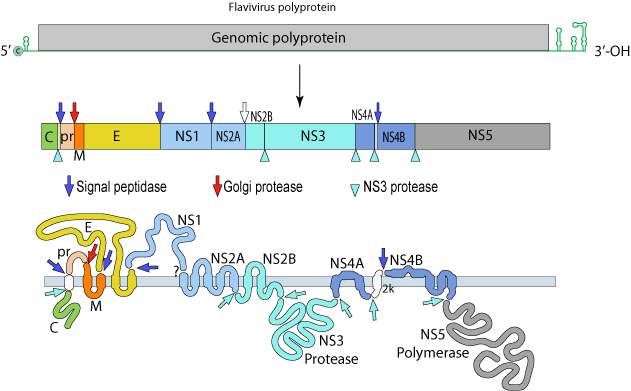
DENV: (+)ssRNA (~11Kb; 1 ORF)
The single polyprotein encodes:
Structural Proteins:
C – capsid
prM – pre-membrane
M - membrane
E - envelope
Non-Structural Proteins:
NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5
Empower the use of HTS to monitor the dissemination of the disease
RNA Extraction
PCR
Amplification
HTS Sequencing
➔
➔
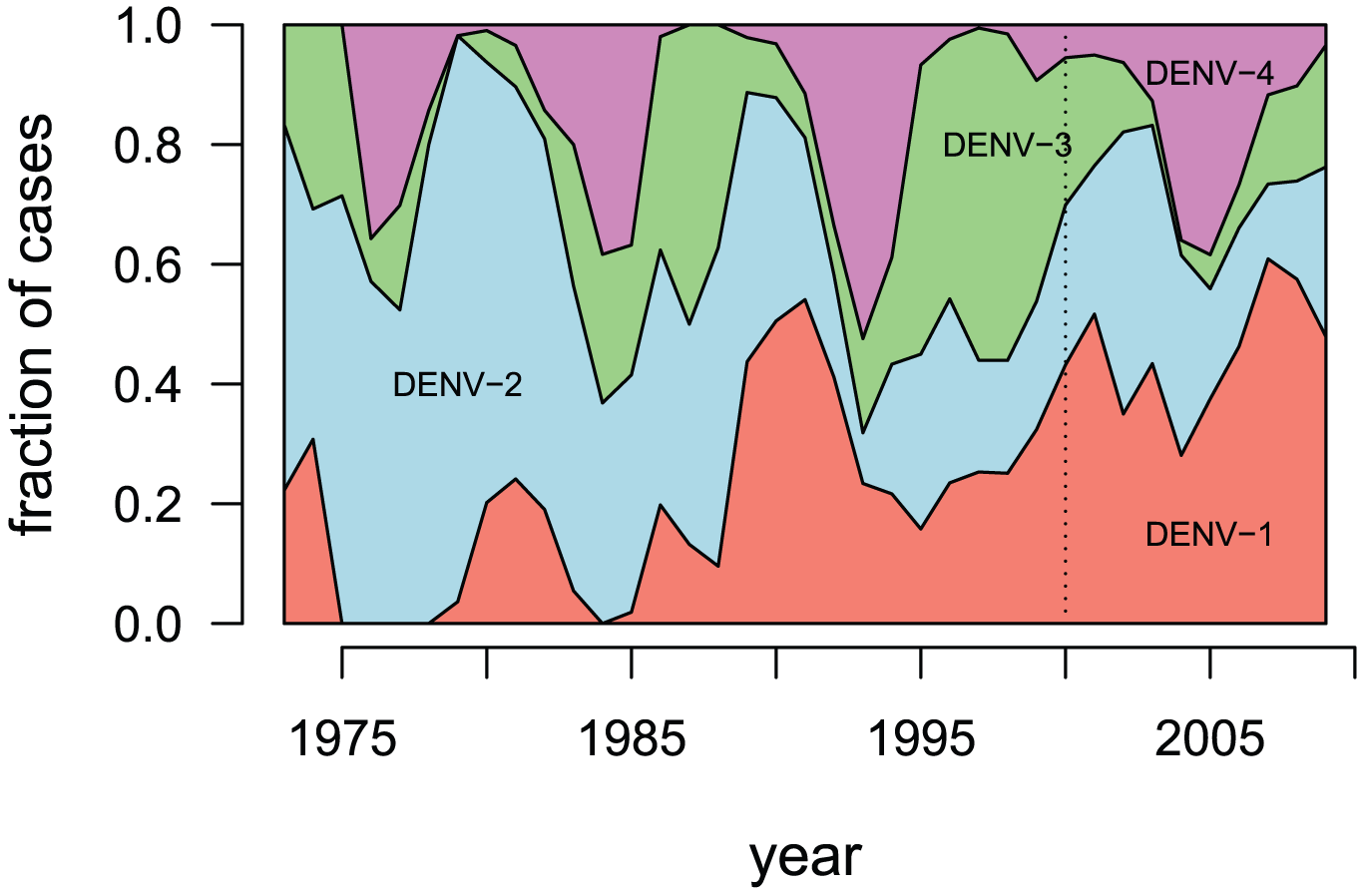
How do the different genotypes model transmission and infection?
Requirements
A solution
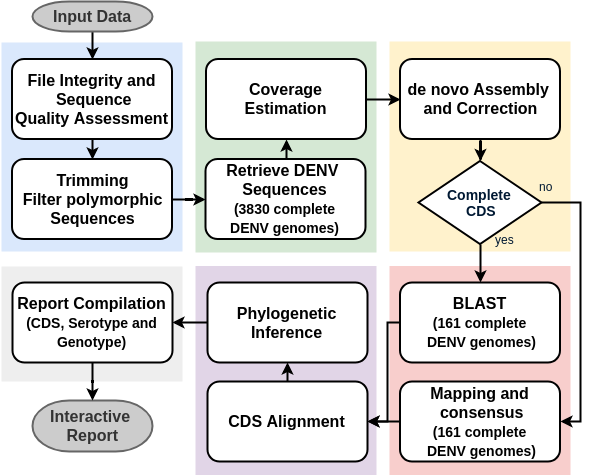
DENV Identification
In Silico Typing:
a) Envelope Region b) whole genome sequence
a) Envelope Region b) whole genome sequence
Shotgun Metagenomics dataset:
nextflow run DEN-IM.nf -profile slurm_shifter --fastq="fastq/*_{1,2}.*"Git, Nextflow (java) and a container engine (Docker, singularity, shifter...).
apt-get install gitcurl -s https://get.nextflow.io | bash
apt-install docker-ceClone (or run remotely)
git clone https://github.com/B-UMMI/DEN-IM.githttps://github.com/B-UMMI/DEN-IM/wikide novo Assembly of short-read data
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
15th of September, 2020
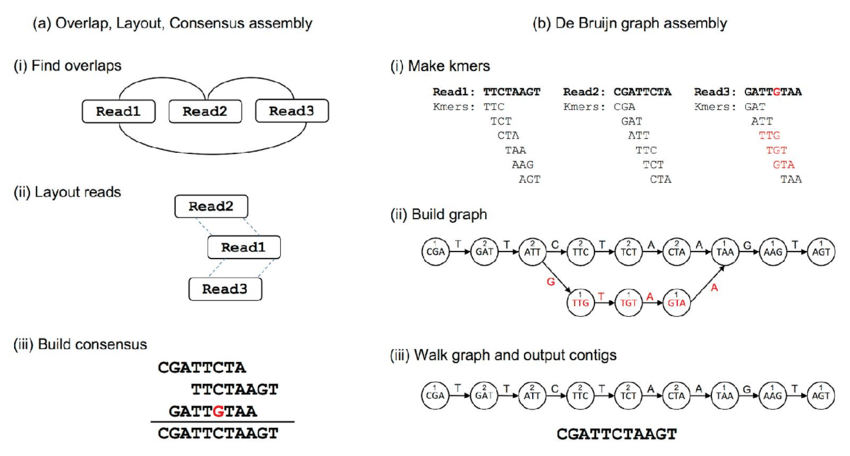
The assembly methods provide longer sequences that are more informative than shorter sequencing data and can provide a more complete picture of the microbial community in a given sample.
Reference Dataset (Complete Bacterial Genomes)
In silico mock sample (even)
In silico mock sample (log)
Zymos standard (even)
Zymos standard (log)
3.7 M read pairs
8.8 M read pairs
47.8 M read pairs
Assembly Workflow
Assembly Quality Assessment
Reference Dataset (Triple)
Assembly file (fasta)
Filter min contig size (1000 bp)
Mapping with Minimpa2
Read Data
PAF file (tab)
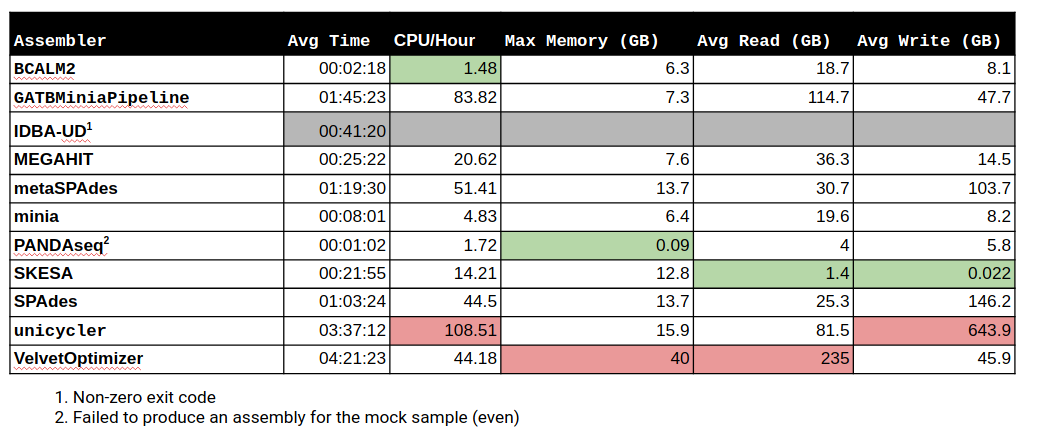
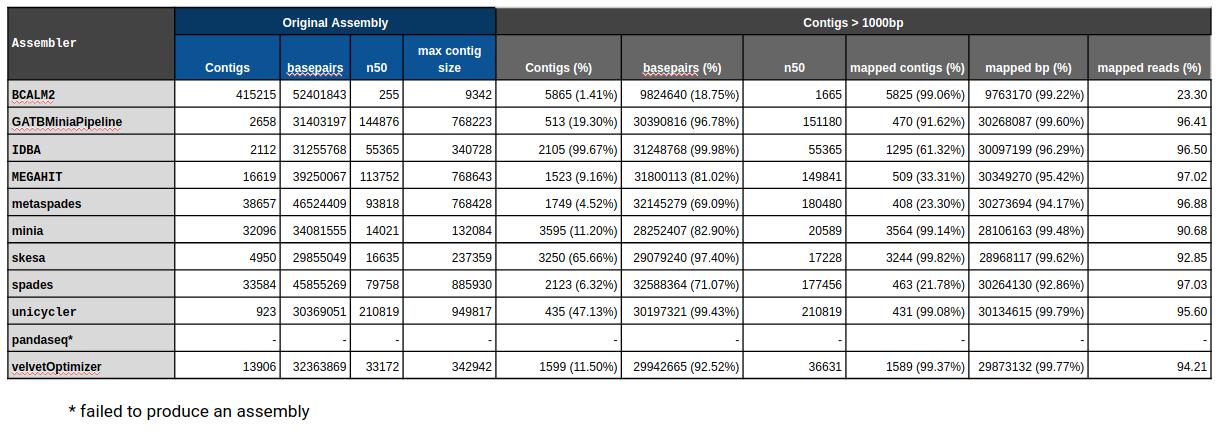
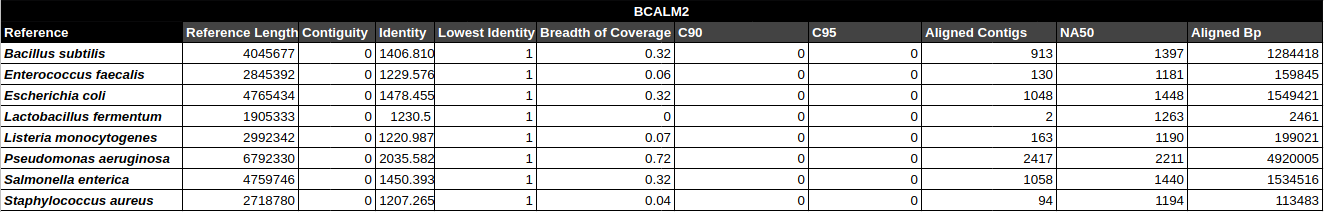
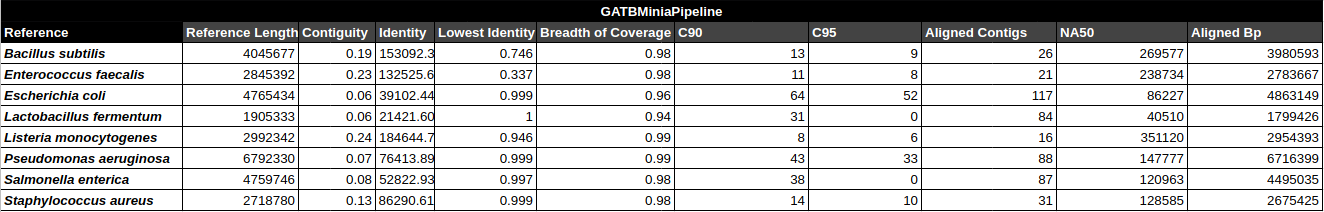
General Assembly & Global Mapping Statistics
Mapping Statistics & Metrics per Reference
Original
Filtered (1000bp)
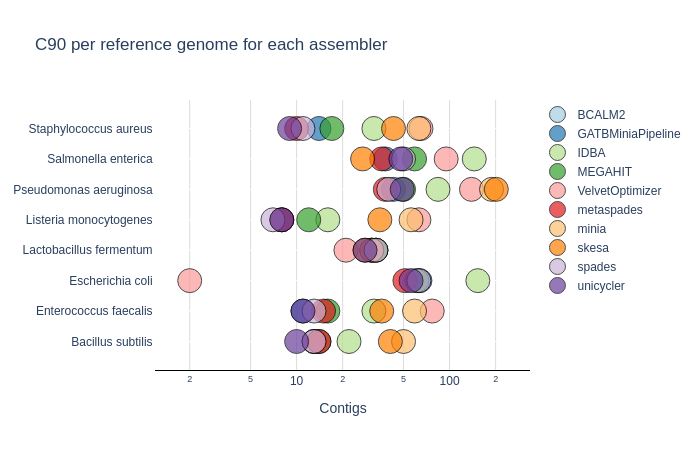
C90 & C95
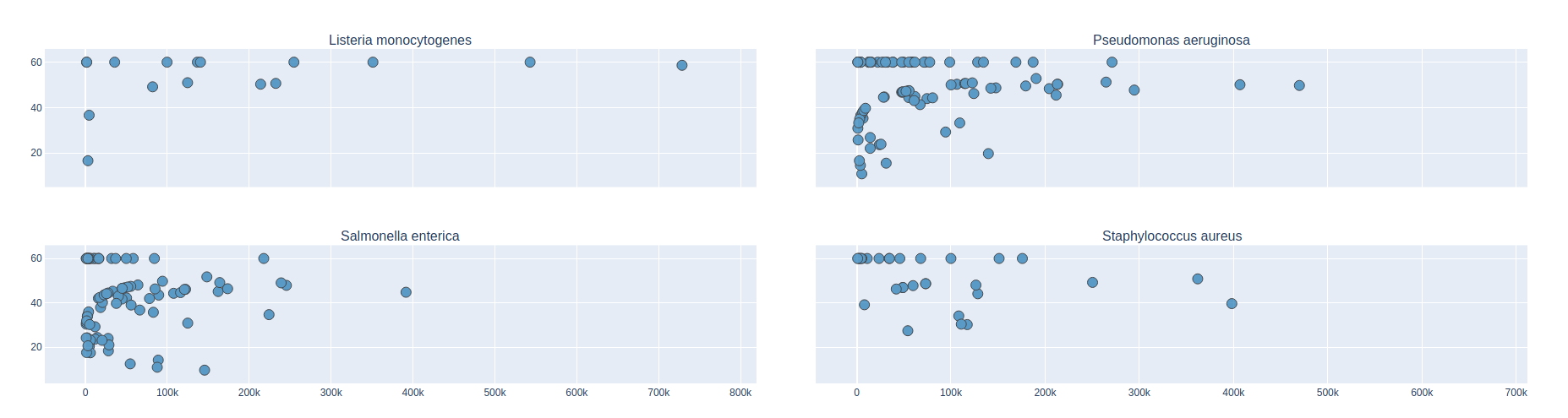
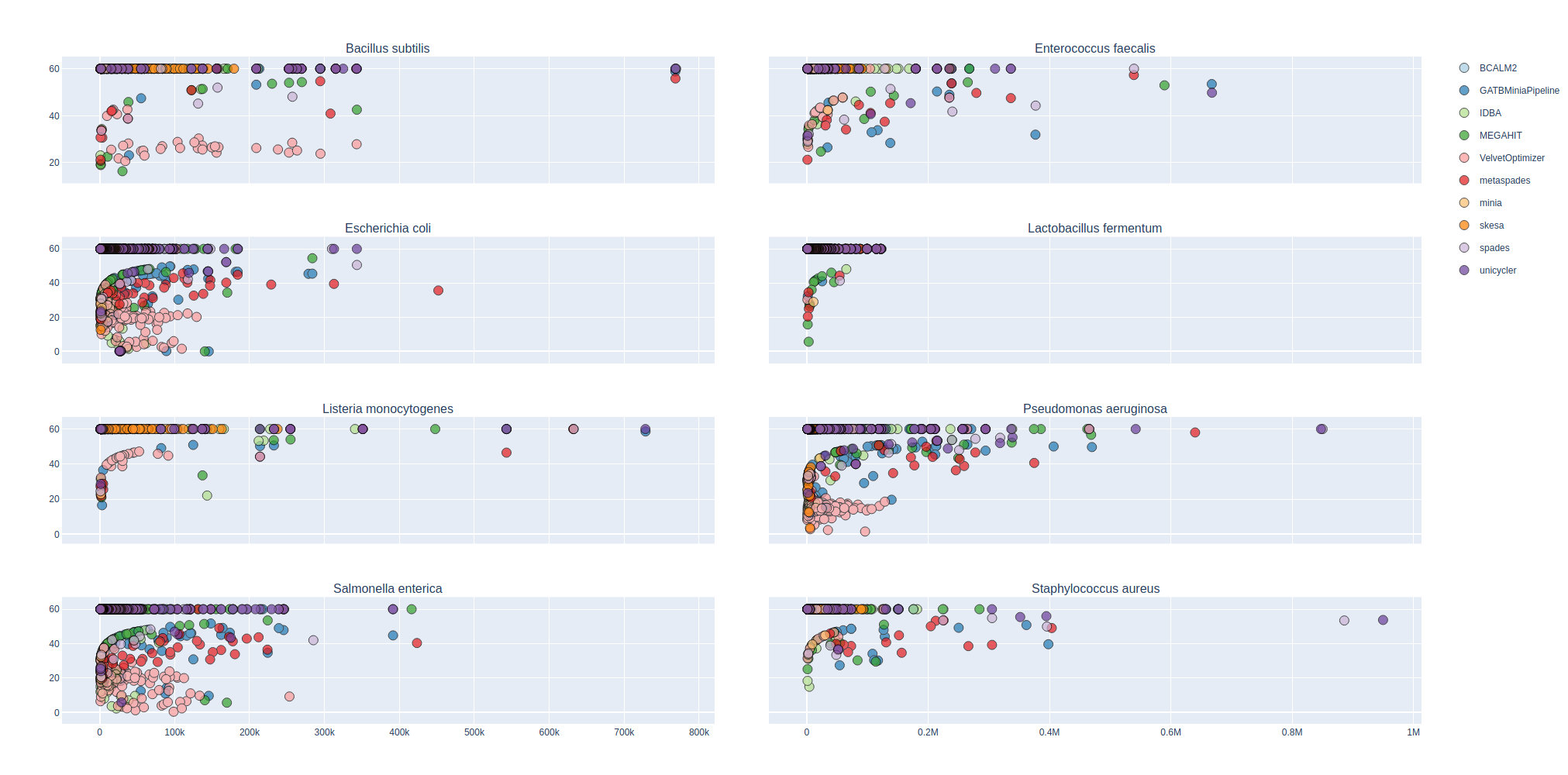
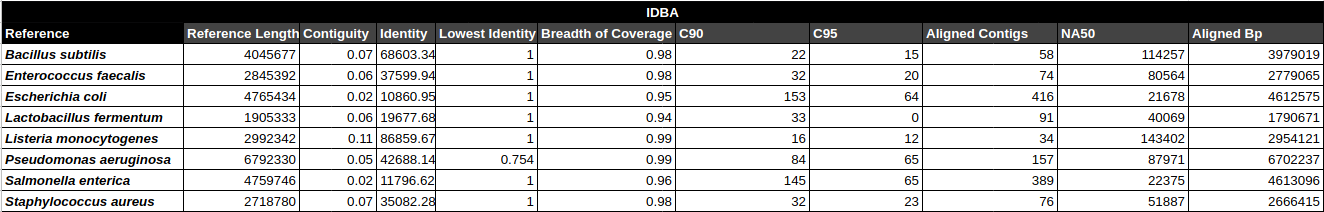
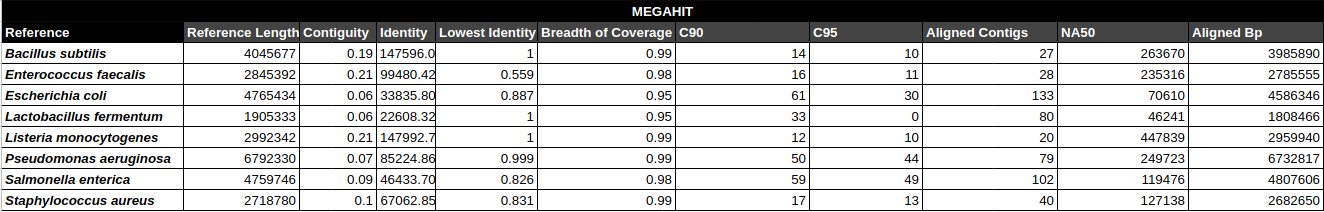
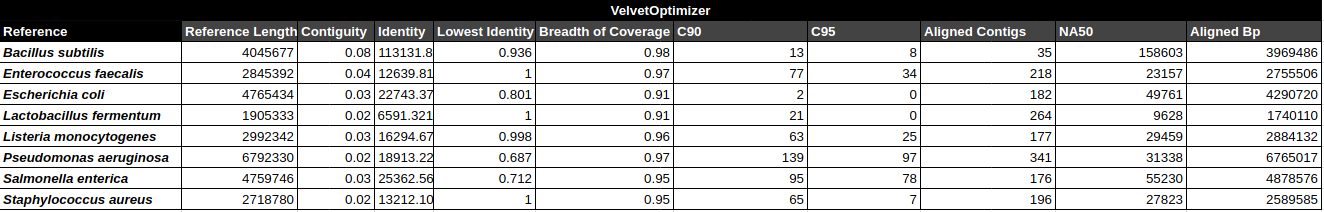
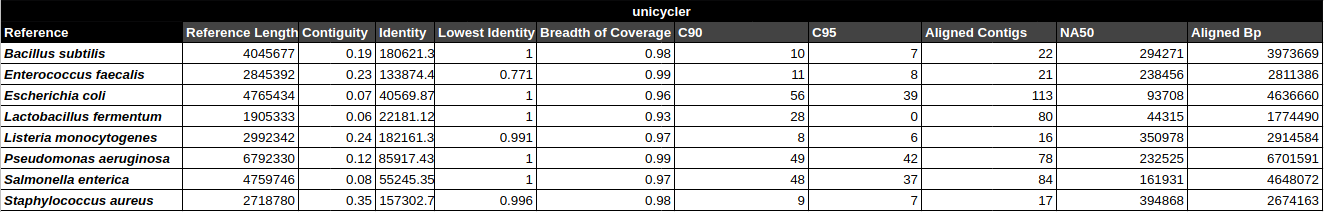
Number of contigs to cover at least 90% and 95% of the reference genome, respectively.
Contig Phread Quality Score
Contiguity
Longest percentage of the reference sequence assembled in a single contig.
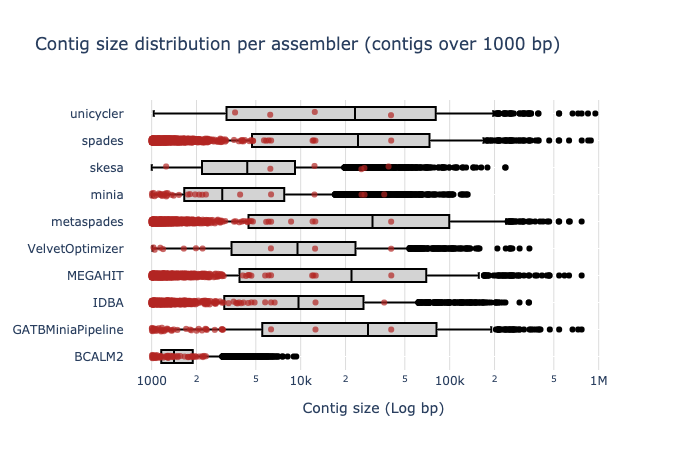
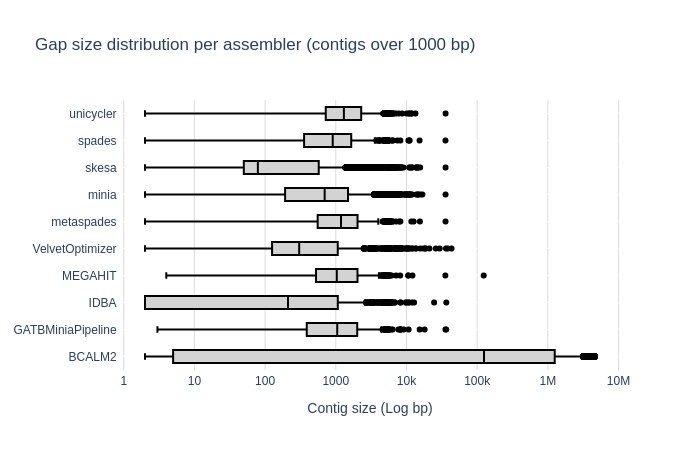
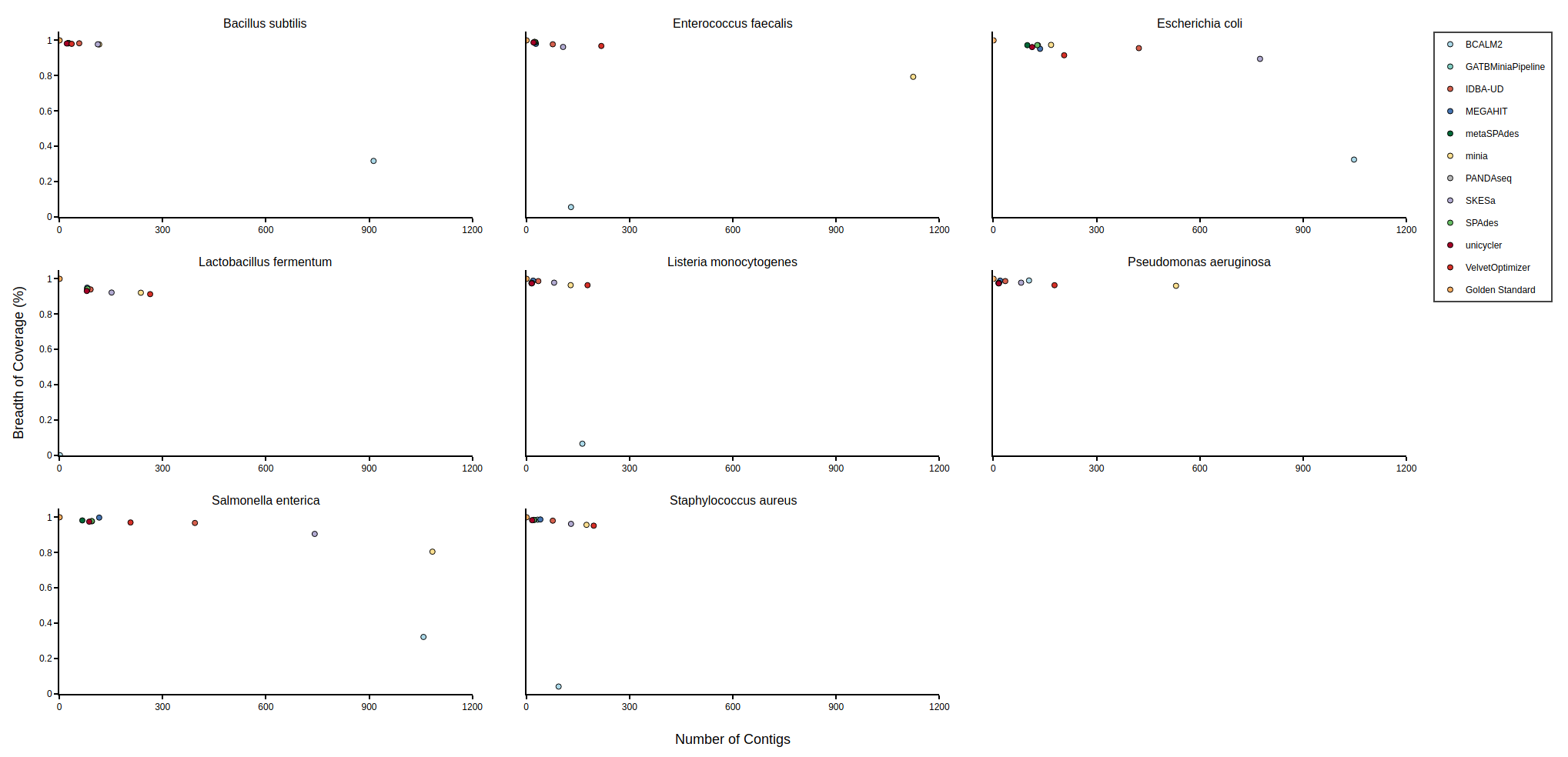
Bradth of coverage and number of contigs per Reference for each Assembler
Contig Phred Quality Score for GATBMiniaPipeline's Pseudomonas aerugiona assemby
Contig Size
Phred Score
Contig Phred Quality Score per Reference for each Assembler
Data Structures & the SARS-CoV-2 Contextual Data Specification
Programa de Doutoramento do Centro Académico de Medicina de Lisboa
Inês Mendes
15th of September, 2020
Standardized data structures and interchangable formats - critical to the development of an open software ecosystem.
Focus on the development, adaptation and standardization of data models for microbial sequence data, contextual metadata, results and workflow metrics to improve the transparency, interoperability and reproducibility of public health sequencing workflows.
Main Goal
SARS-CoV-2 contextual data specification that incorporates publicly available community standards, as well as additional fields and guidance appropriate for public health surveillance and analyses.
Resources
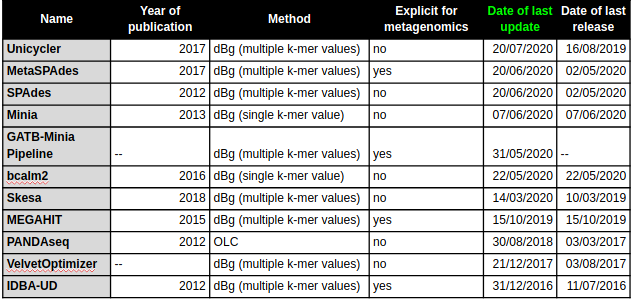
Continue benchmark analysis for the mock semple (log distributed) and the real samples (Zymos community standards log and evenly distributed).
PHA4GE - Harmonization of tool outputs for the detection of antimicrobial registance genes (https://github.com/pha4ge/hAMRonization).
Web service for the interactive vizualization of Kraken's taxonomic composition reports.
Nothing else Meta - Reference indenpendent filtration of human reads from (meta)genomic datasets.
Special thanks to Diogo Silva, Bruno Gonçalves, Tiago Jesus, Pedro Vila-Cerqueria, Rafael Maria Mamede, João Carriço, John Rossen and Mário Ramirez.
By Inês Mendes
CAML PhD Program Thesis Comittee - 15 September 2020




