
















Inês Mendes
Bioinformatics PhD student.
Inês Mendes
MRamirez Lab - iMM
@ines_cim
cimendes
Metagenomic Assembly
Towards Accreditation in Metagenomics for Clinical Microbiology
Advisor Comittee Meeting
14 July 2021
Shotgun Sequencing Data
| Assembly
The assembly methods provide longer sequences that are more informative than shorter sequencing data and can provide a more complete picture of the microbial community in a given sample.
Assembly
consensus
de novo
OLC
de Bruijn graph
| Assembly
Major issues
Reads
Contigs
Genomes
| Assembly
Main goals
Reads
Contigs
Genomes
| Assembly
Main limitations
Reads
Contigs
Genomes
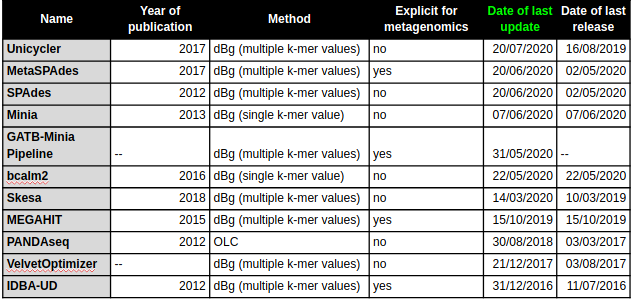
Compiled a collection of de novo assembly tools, including Overlap, Layout and Consensus (OLC) and De Bruijn graph (dBg) assembly algorithms, with both single k-mer and multiple k-mer value approaches, and hybrid assemblers. The collection includes both genomic and metagenomic assemblers.
Initial development of global and reference specific performance metrics for the assessment of de novo assembly quality.
https://github.com/cimendes/metagenomic-assembler-comparison
Reference Dataset (Complete Bacterial Genomes)
In silico mock sample (even)
In silico mock sample (log)
Zymos standard (even)
Zymos standard (log)
3.7 M read pairs
8.8 M read pairs
47.8 M read pairs
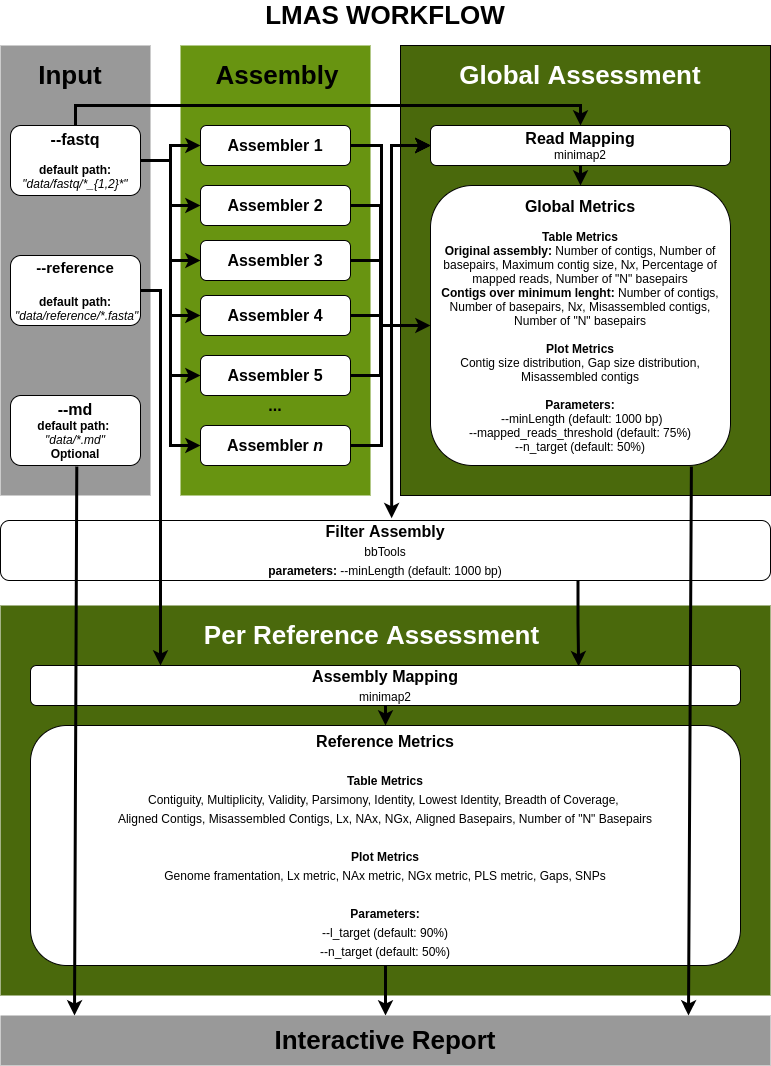
Basic Assembly Workflow
Assembly Quality Assessment
https://github.com/cimendes/LMAS
https://lmas.readthedocs.io/
| Last Metagenomic Assembler Standing
Automated workflow enabling the benchmarking of genomic and metagenomic prokaryotic de novo assembly software using defined mock communities.
| Last Metagenomic Assembler Standing
The input data is assembled in parallel by the set of genomic and metagenomic de novo assemblers in LMAS.
The global and per reference metrics are grouped in the interactive LMAS report for exploration.
The resulting assembled sequences are processed and assembly quality metrics are computed.
| Last Metagenomic Assembler Standing
| Last Metagenomic Assembler Standing
LMAS requires a Nextflow installation (version ≥ 21.04.1), requiring BASH and Java 8 (or higher).
All components of LMAS are executed in containers.
Continuous integration of the python templates for the quality assessment of assemblies is performed with pytest and GitHub Actions.
LMAS can be installed through Github.
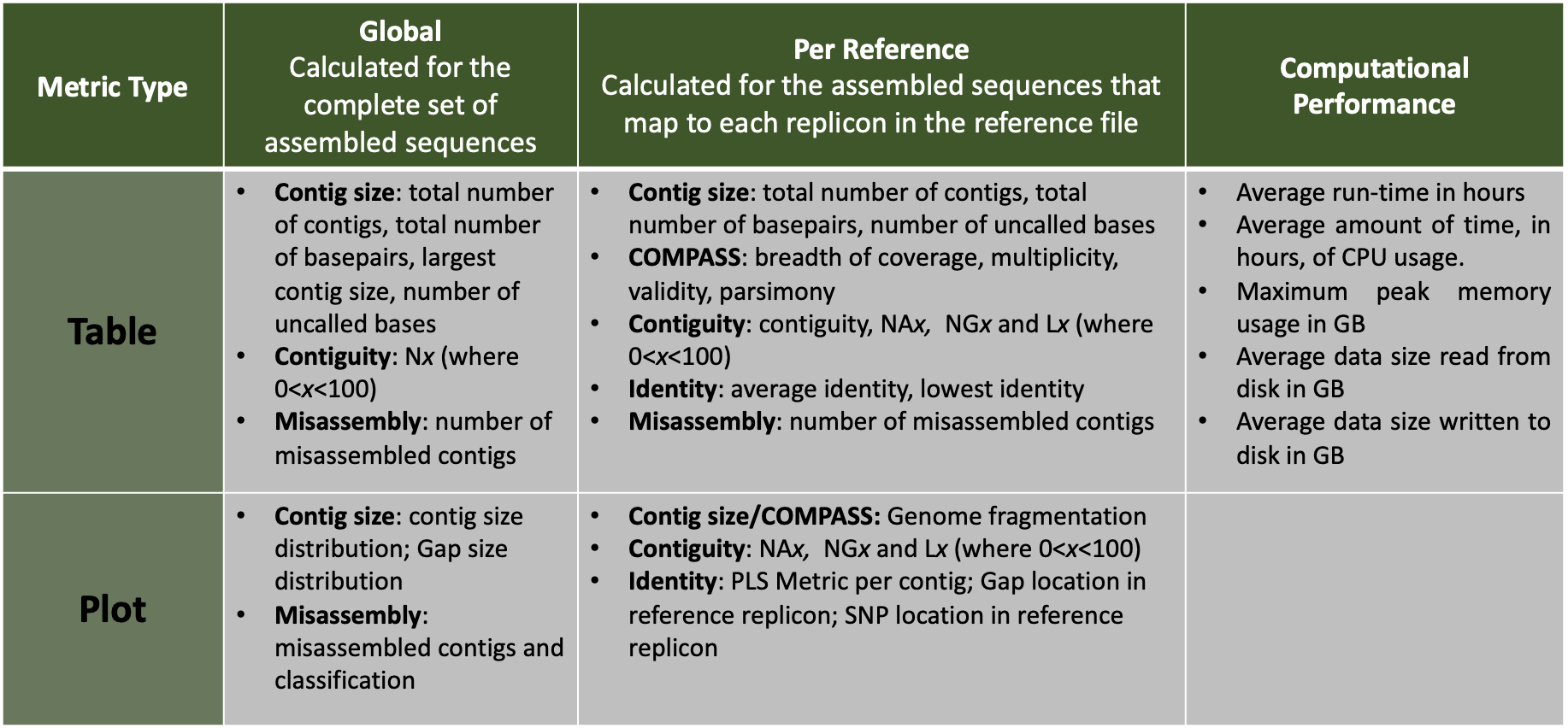
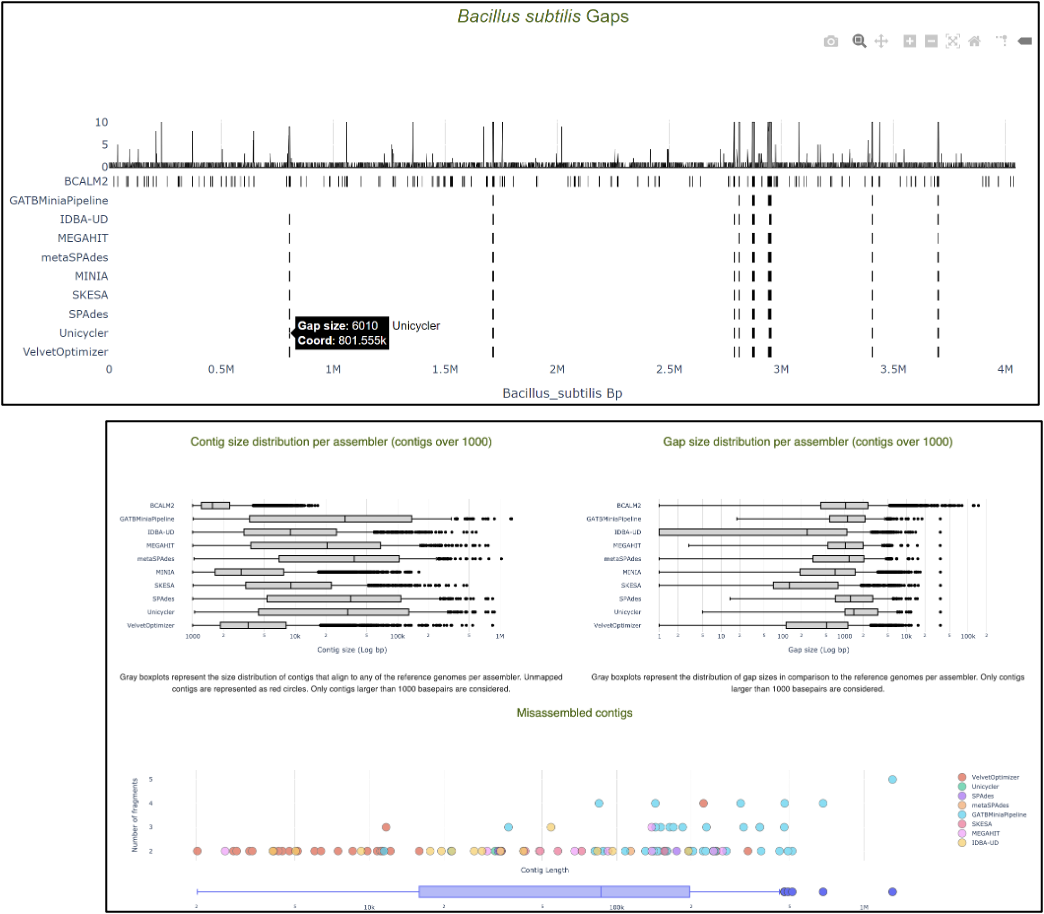
| Assembly Quality Metrics
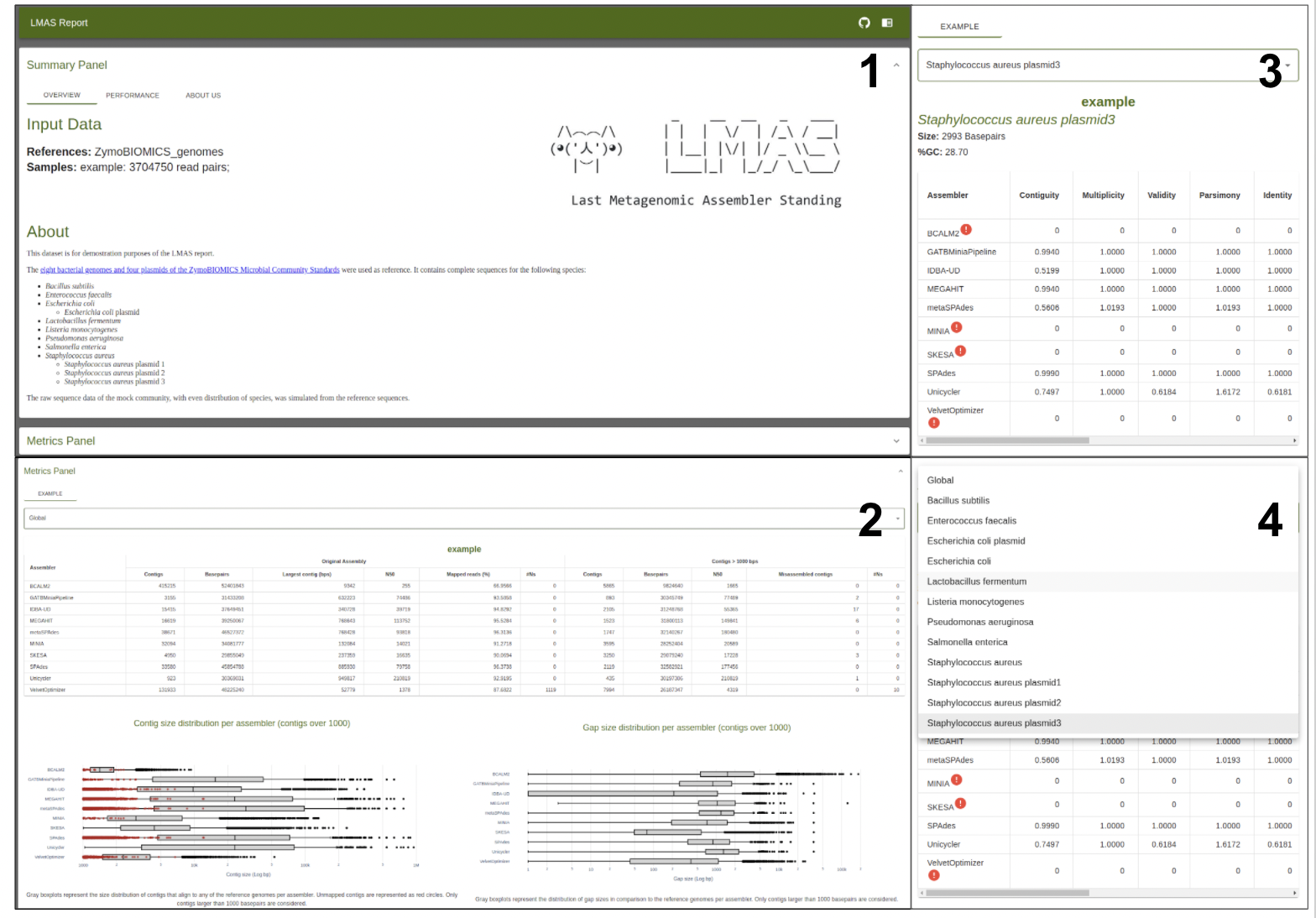
The tabular presentation allows direct comparison of exact values between assemblies, and the interactive plots allow for an intuitive overview and easy exploration of results.
| Report
| Report
The results are presented in an interactive HTML composed of two main panels:
- a top summary panel with information on input samples and the LMAS execution,
- a bottom panel where selected global and reference specific performance metrics can be explored for each sample.
The JavaScript source code for the interactive report comes bundled with LMAS but is freely available at https://github.com/cimendes/lmas_report.
| Sample | Distribution | Error Model | Read Pairs (M) |
|---|---|---|---|
| ENN | Even | None | 8.6 |
| EHS | Even | Illumina HiSeq | 8.6 |
| ERR2984773 | Even | Real Sample | 8.6 |
| LNN | Log | None | 47.5 |
| LHS | Log | Illumina HiSeq | 47.5 |
| ERR2935805 | Log | Real Sample | 47.5 |
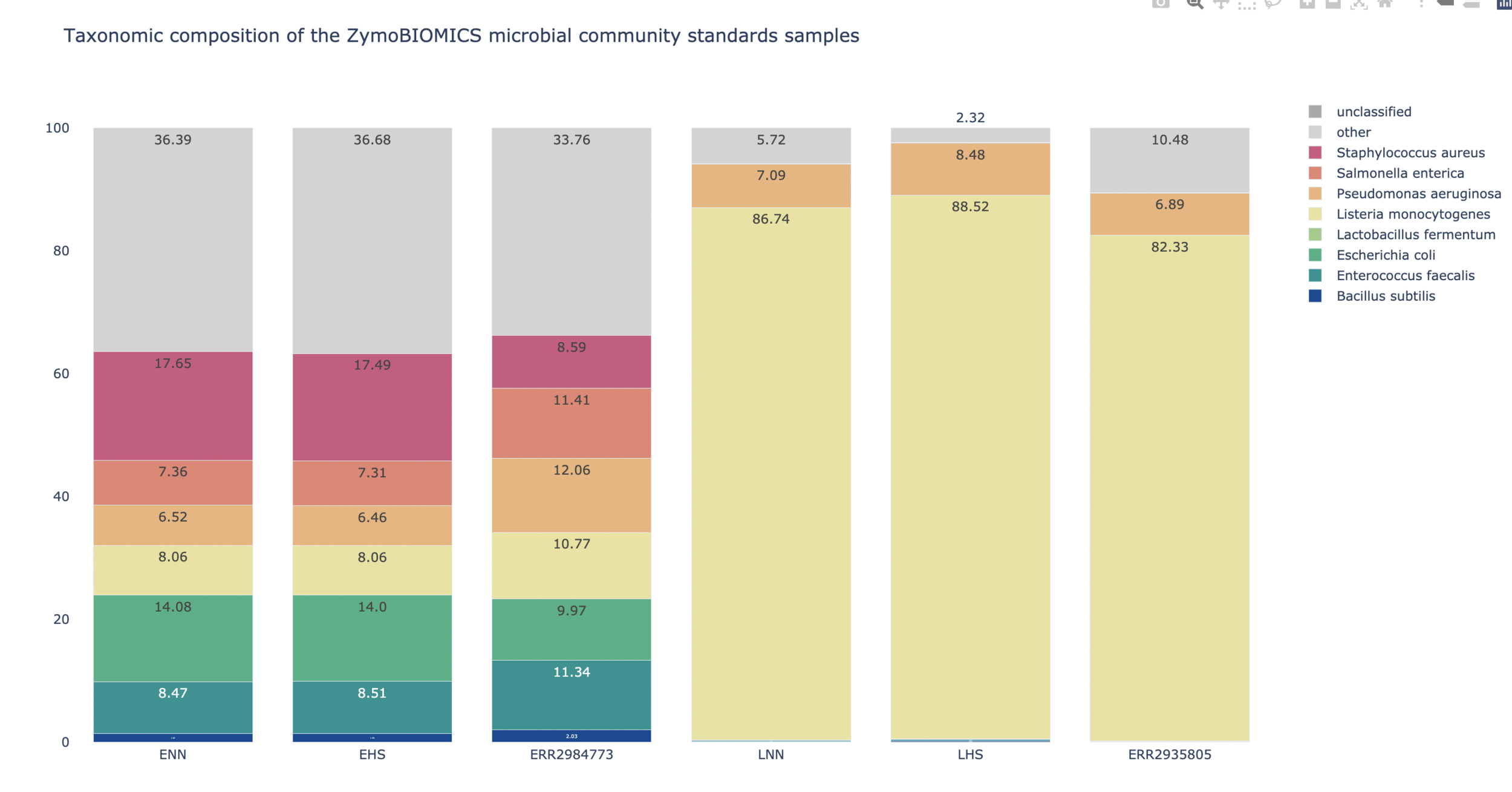
| ZymoBIOMICS Microbial Community Standards
| ZymoBIOMICS Microbial Community Standards
| ZymoBIOMICS Microbial Community Standards
No one size fits all assembler!
| 3 LMAS runs
https://github.com/cimendes/LMAS_Zymos_Resuls
| 3 LMAS runs
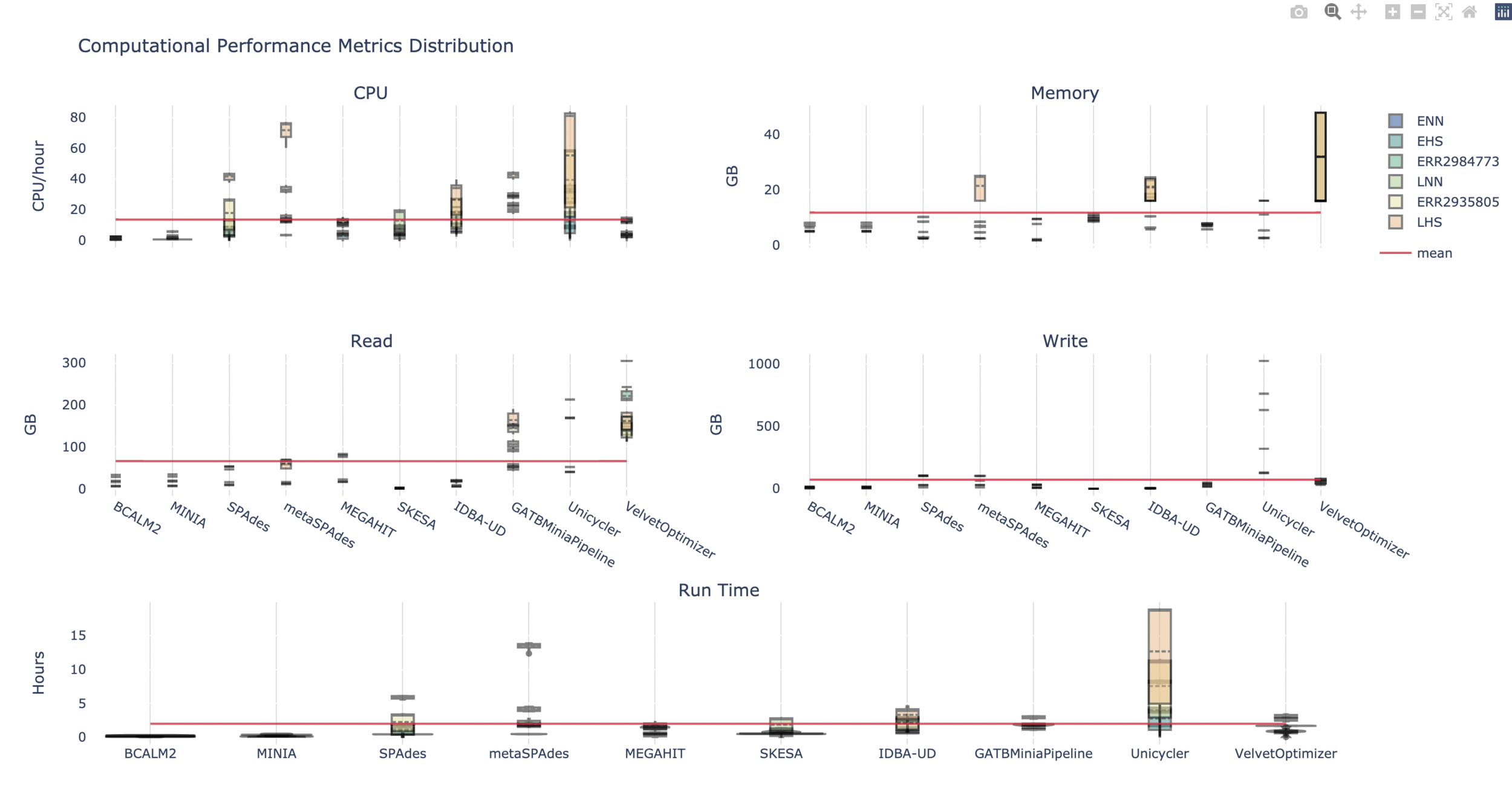
There’s a disparity in usage for the evenly and logarithmically distributed samples, with the latter having more resource-intensive requirements. The resource usage also varied greatly by assembler, with multiple k-mer dBG having overall higher resource usage.
| 3 LMAS runs
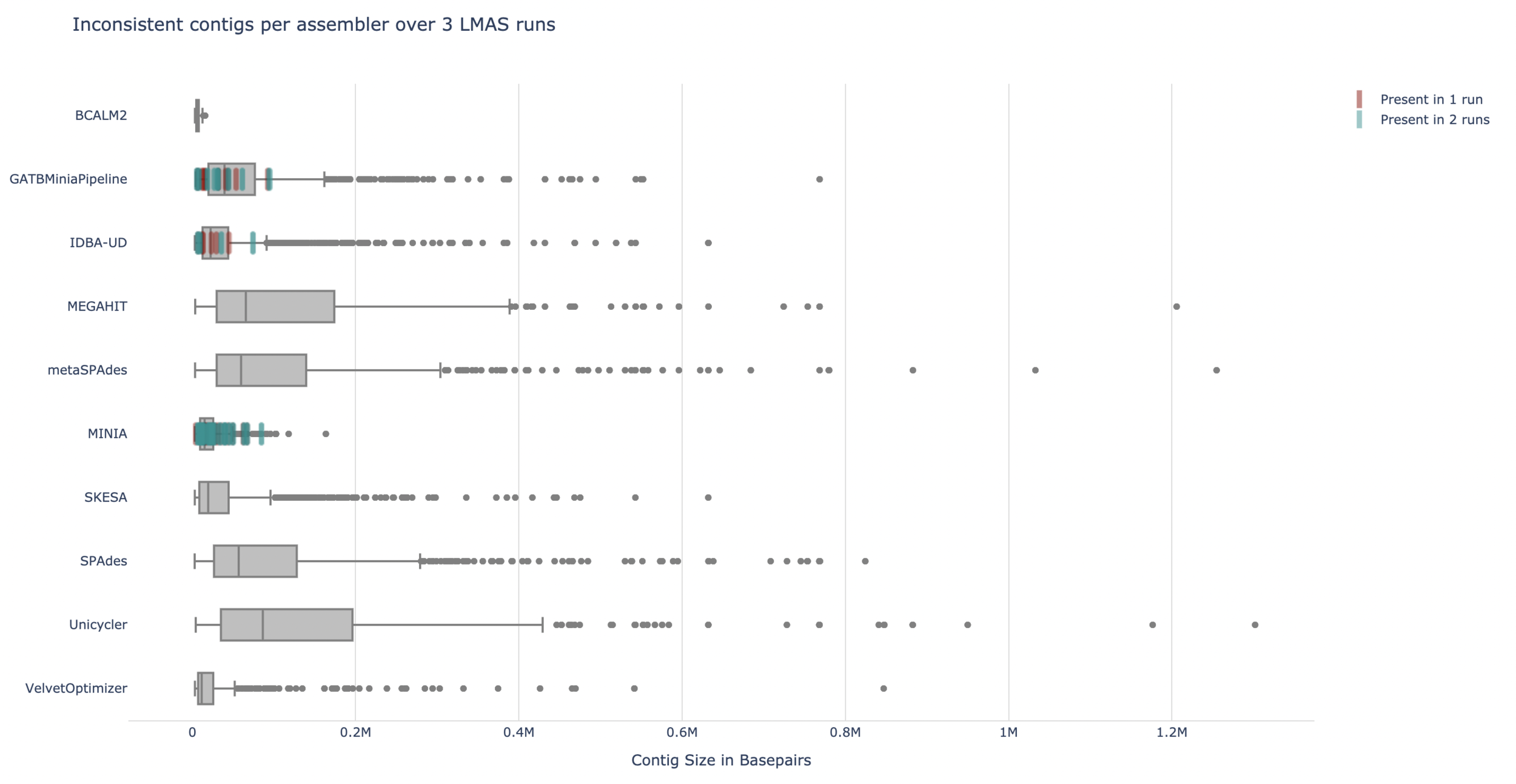
Only GATBMiniaPipeline, IDBA-UD and Minia assemblers produced inconsistent contigs (0.22%, 0.04% and 0.12% of the total contigs produced by the assemblers, respectively)
| 3 LMAS runs
| Know issues
Generate mock dataset with matching read number and sample abundance distribution https://github.com/HadrienG/InSilicoSeq/issues/208
| Know issues
| Nice to have
Allow for long and hybrid assembly https://github.com/cimendes/LMAS/tree/dev_long_read
| Nice to have
Allow for long and hybrid assembly https://github.com/cimendes/LMAS/tree/dev_long_read
By Inês Mendes




