









Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/stanford2023
I am on the scientific advisory boards of Apriori Bio, Aerium Therapeutics, Invivyd, and the Vaccine Company.
I am an inventor on Fred Hutch licensed patents related to deep mutational scanning of viral proteins
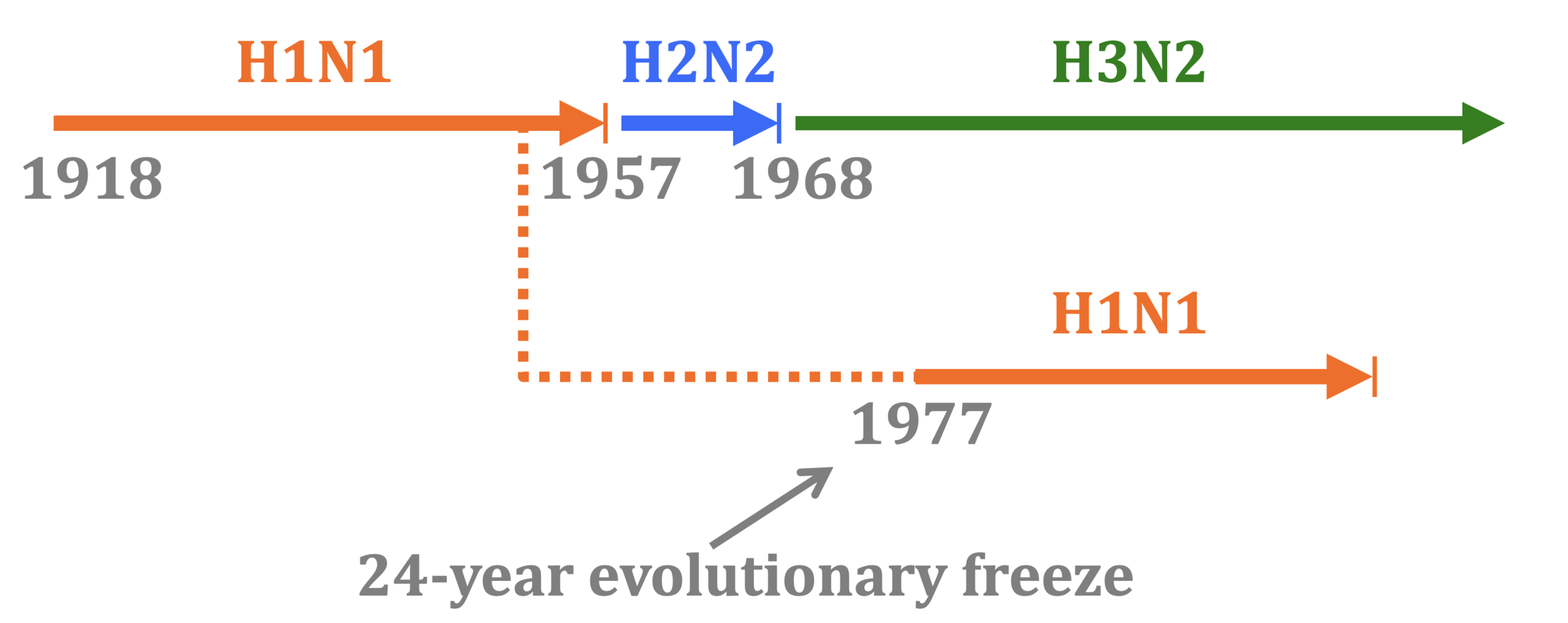
In 1977, old H1N1 strain from ~1954 was inadvertently re-released and caused a global pandemic.
"One boy from Hong Kong had a transient febrile illness from 15 to 18 January. On Sunday 22 January, three boys were in the college infirmary… 512 boys (67%) spent between three and seven days away from class."
"Of about 130 adults who had some contact with the boys, only one, a house matron, developed similar symptoms."
Why some viruses evolve to escape immunity while others don't is a deep question outside scope of this talk. See here for some possible explanations.
Rate of viral antigenic evolution
Measles
Influenza
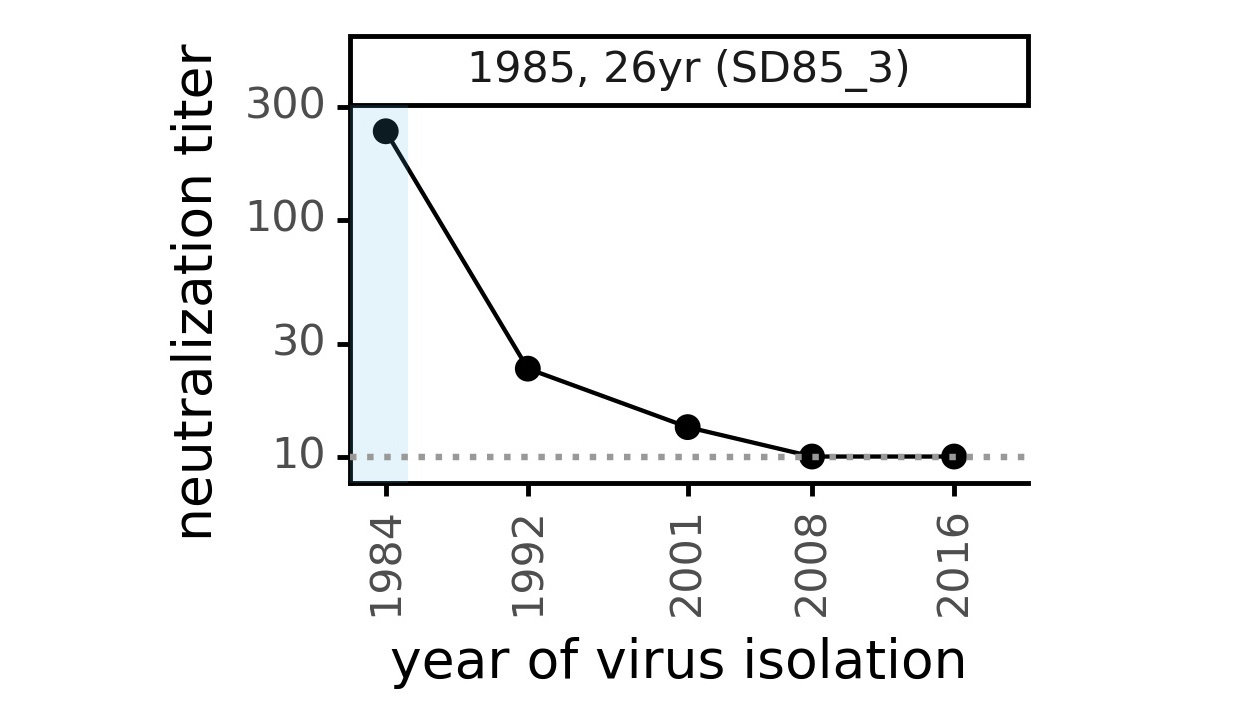
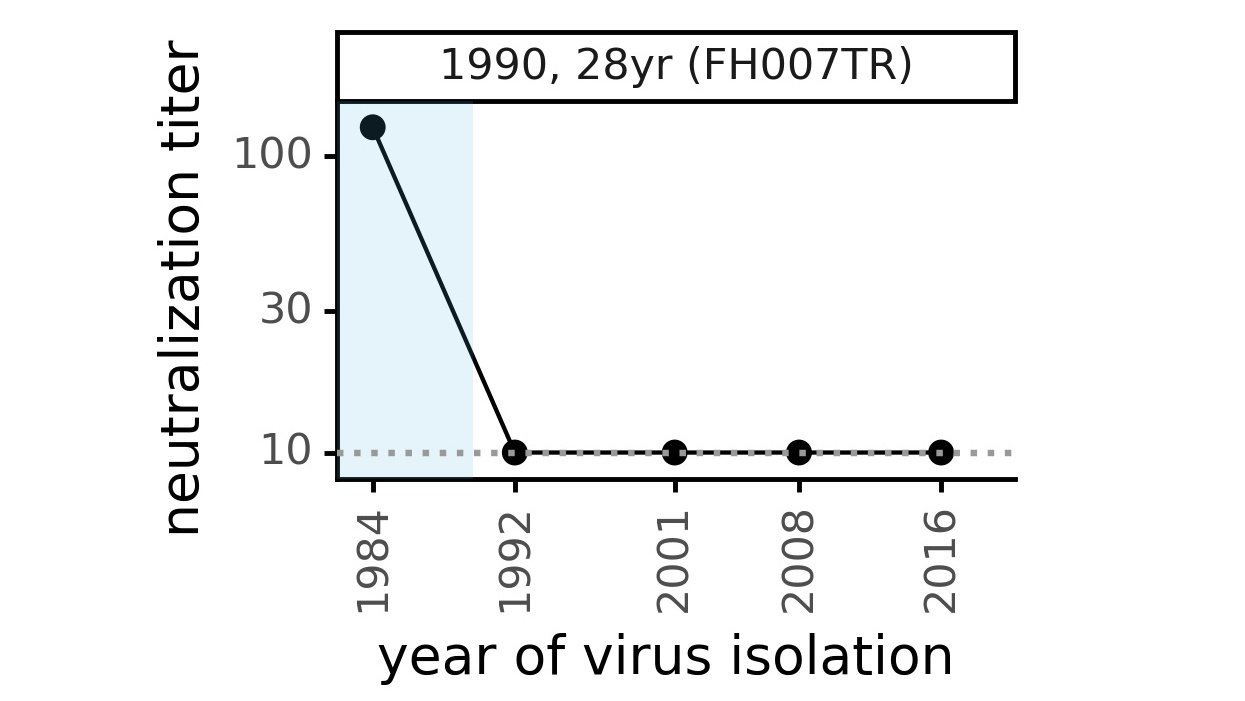
CoV-229E causes common colds and has been circulating in humans for a long time.
The typical person is infected every ~3 to 5 years.
We experimentally generated CoV-229E spikes at ~8 year intervals so we could study them in the lab:
- 1984
- 1992
- 2001
- 2008
- 2016
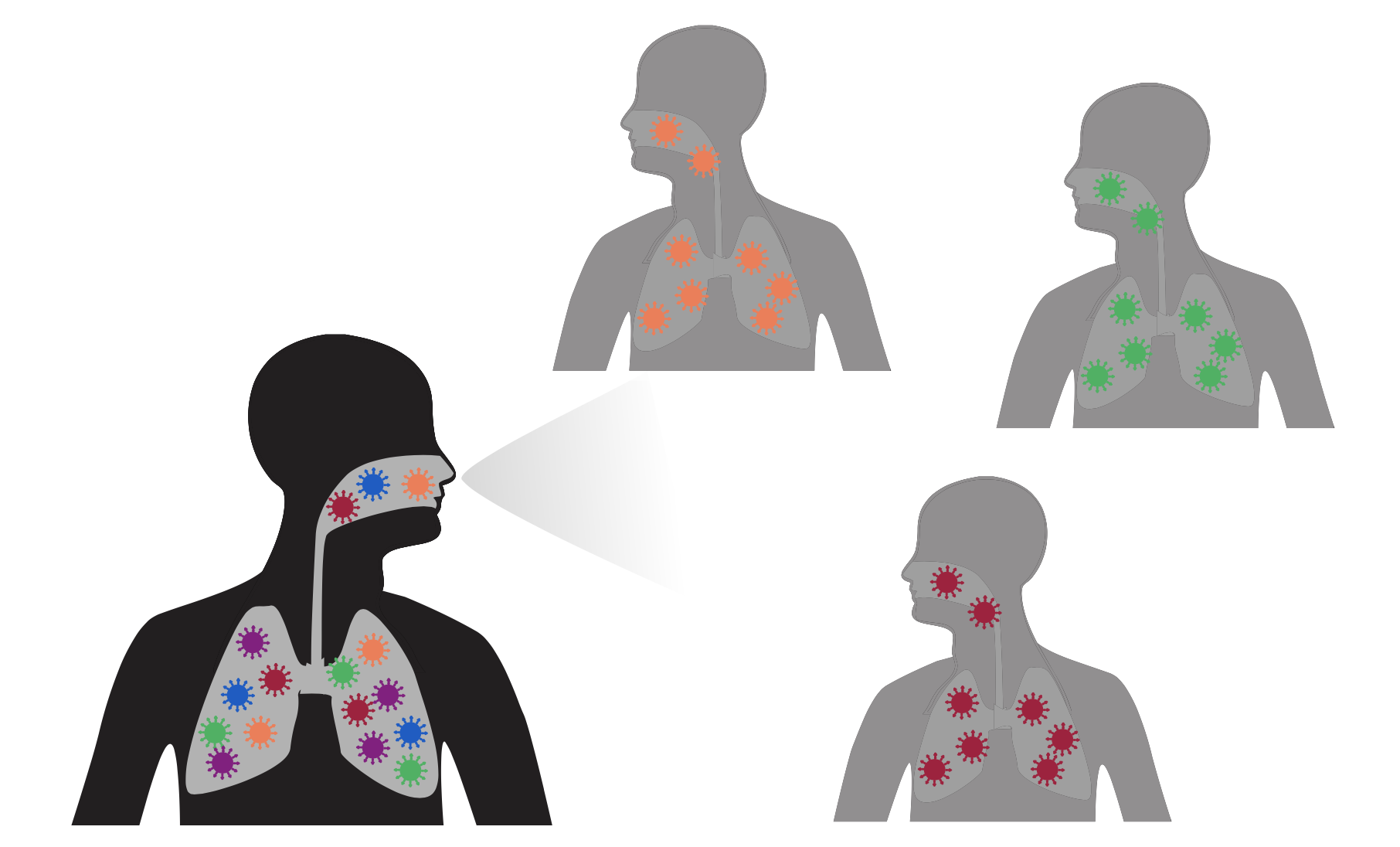
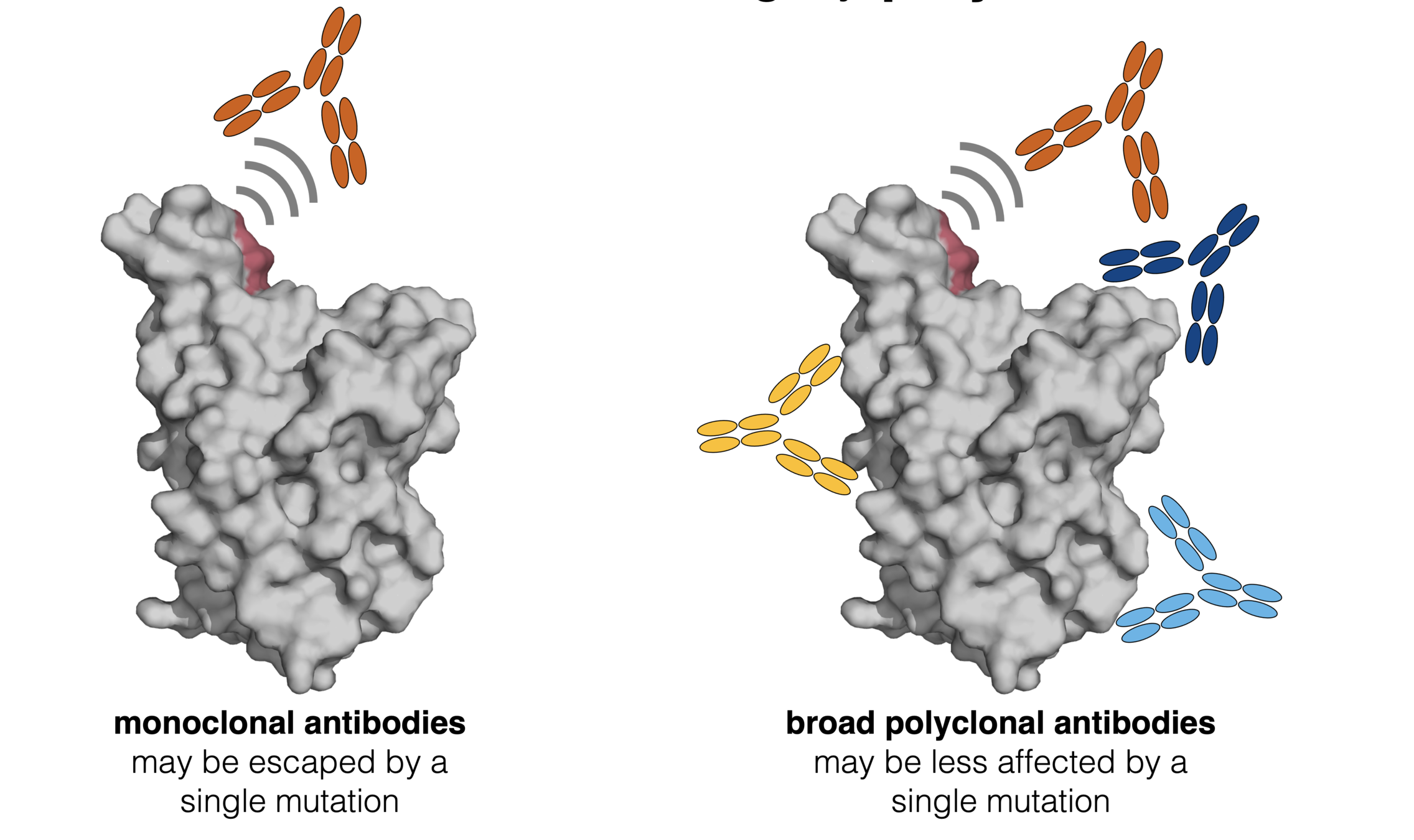
Ideally vaccines would elicit evolution-resistant neutralizing antibodies (like those naturally made by person at right) rather than evolution-sensitive antibodies (like those naturally made by person at left)
Sites of evolutionary change in the spike of CoV-229E over the last four decades
Sites of evolutionary change in the spike of CoV-229E over the last four decades
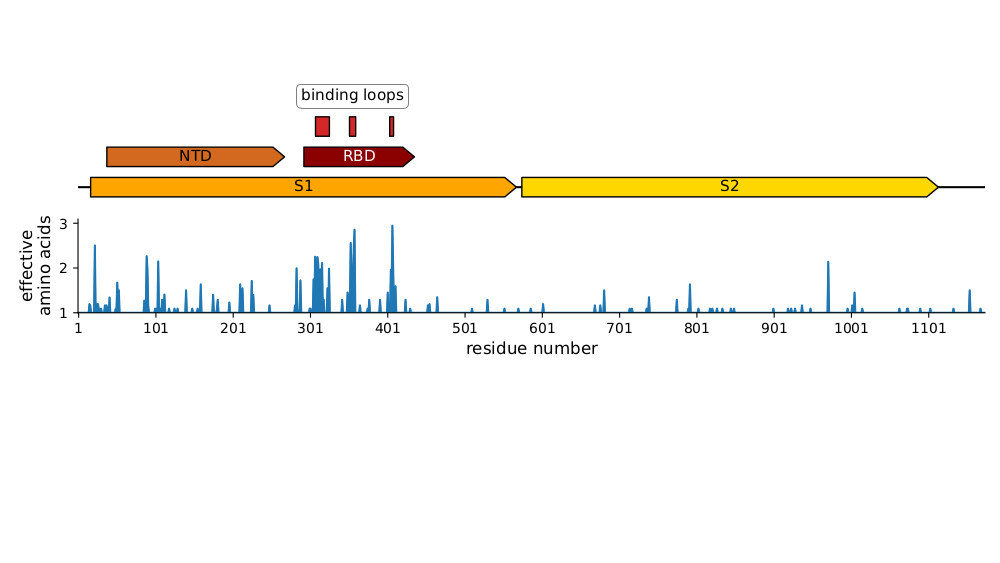
Sites of mutations in SARS-CoV-2 Omicron BQ.1.1 spike relative to Wuhan-Hu-1
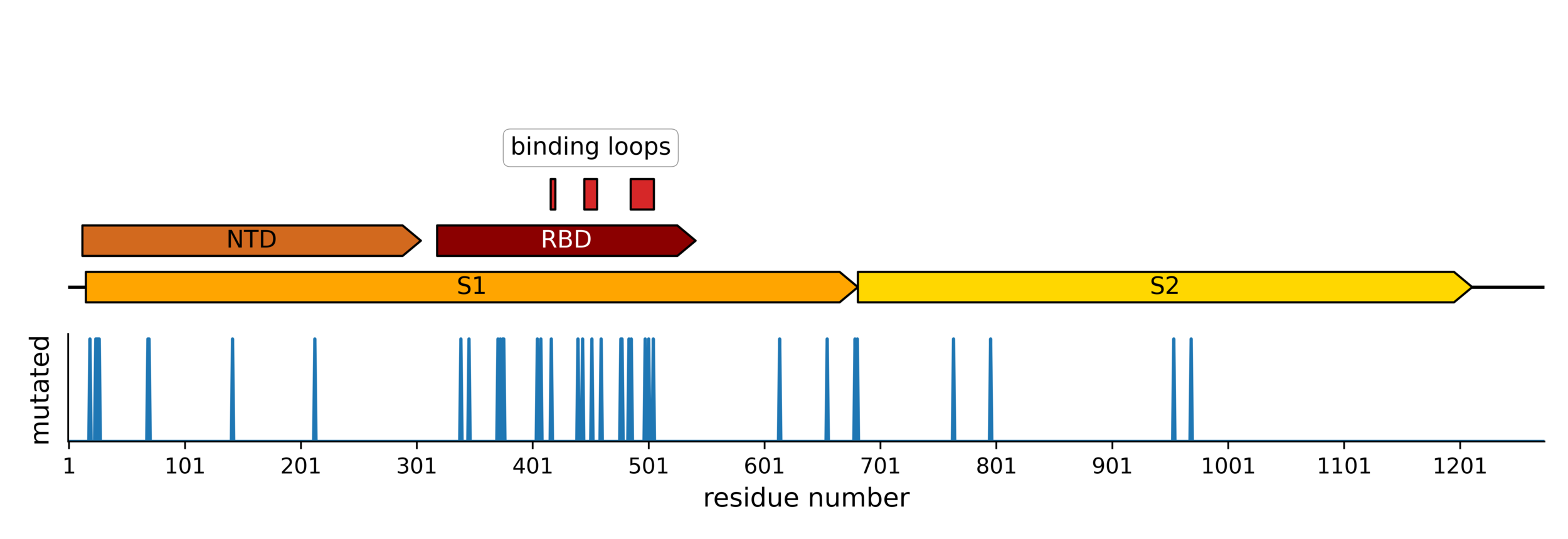
25 of 31 residues in CoV-229E RBD that contact receptor varied during virus's evolution in humans over last ~50 years (Li et al, 2019)
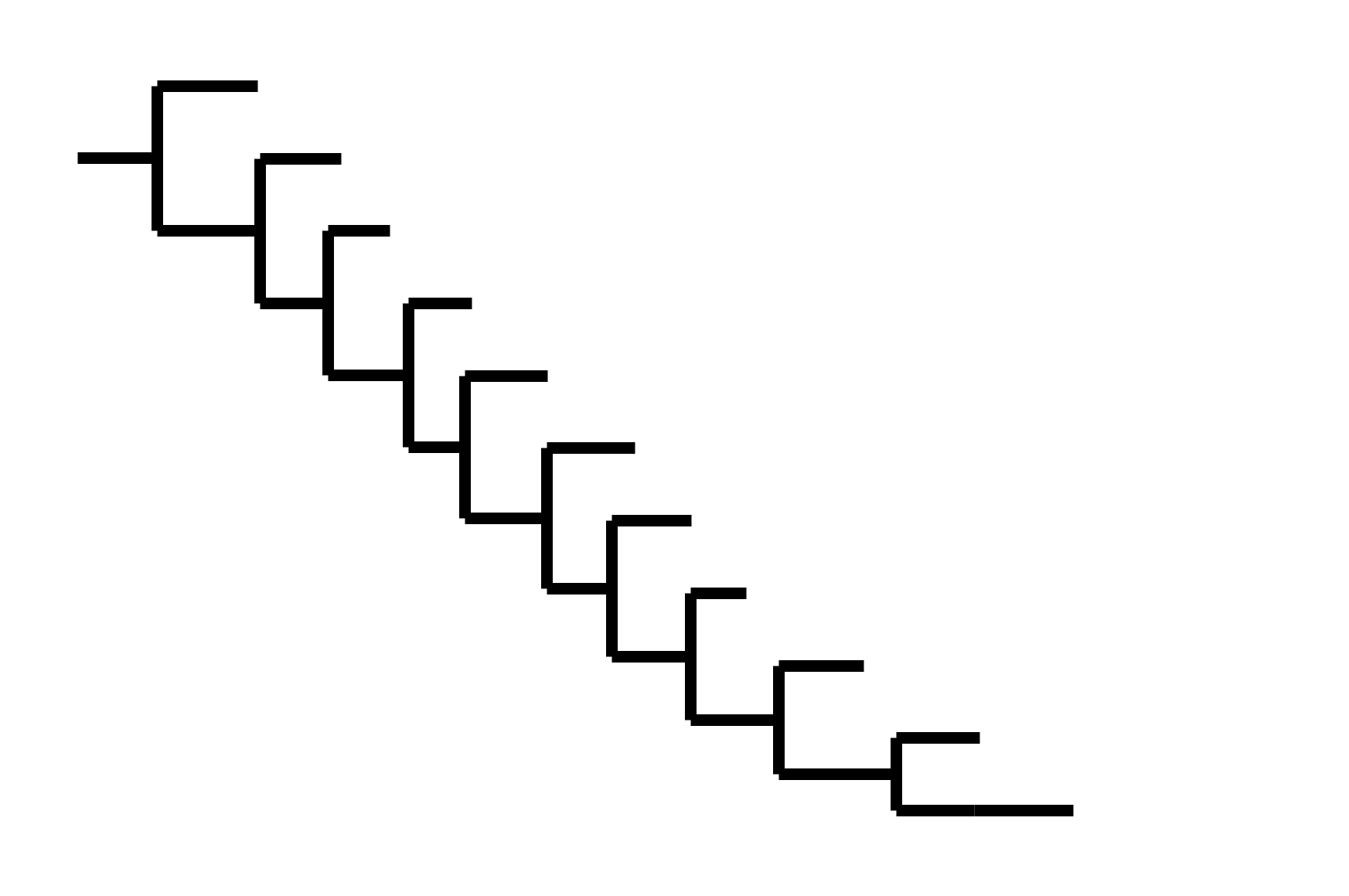
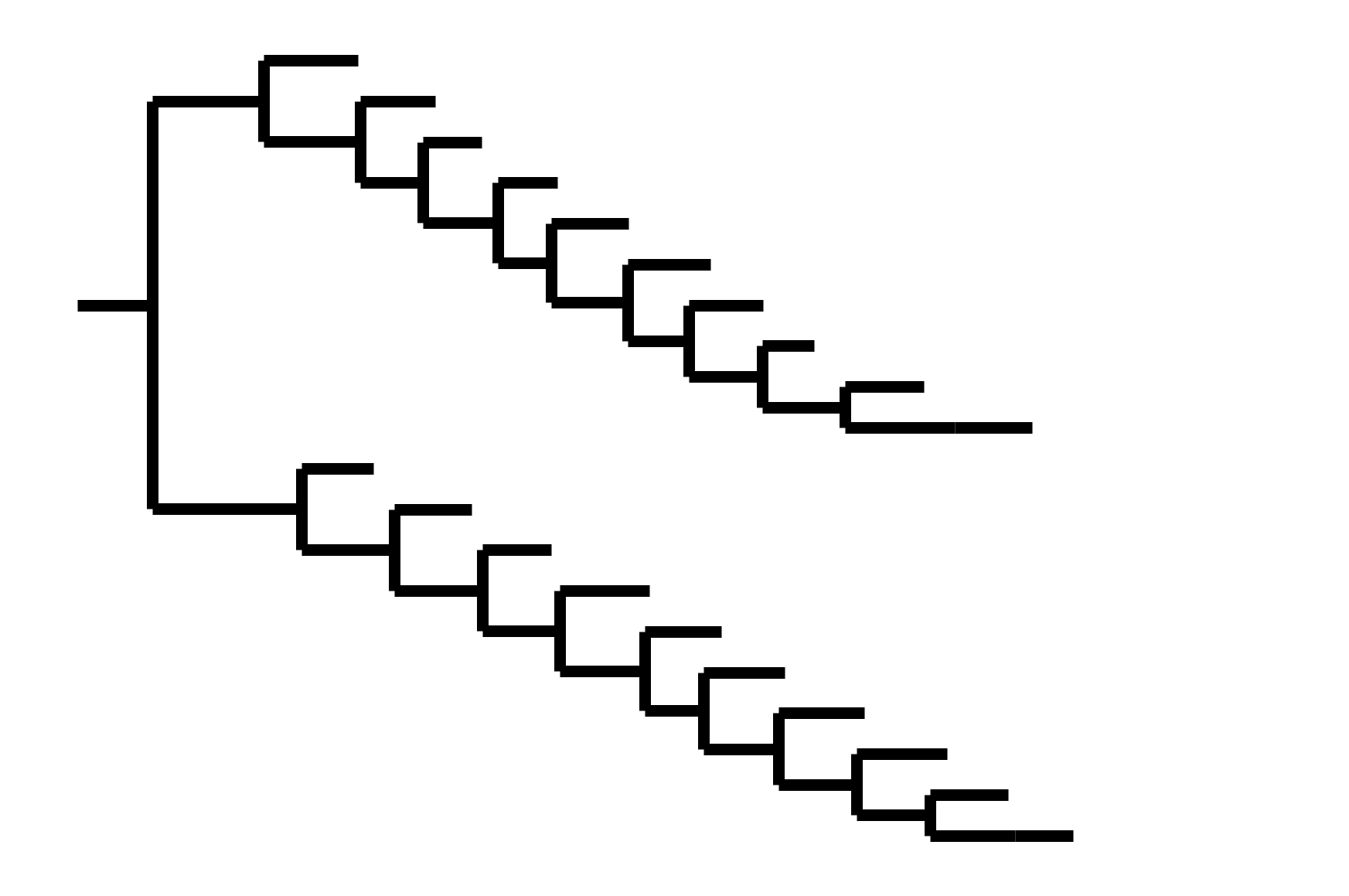
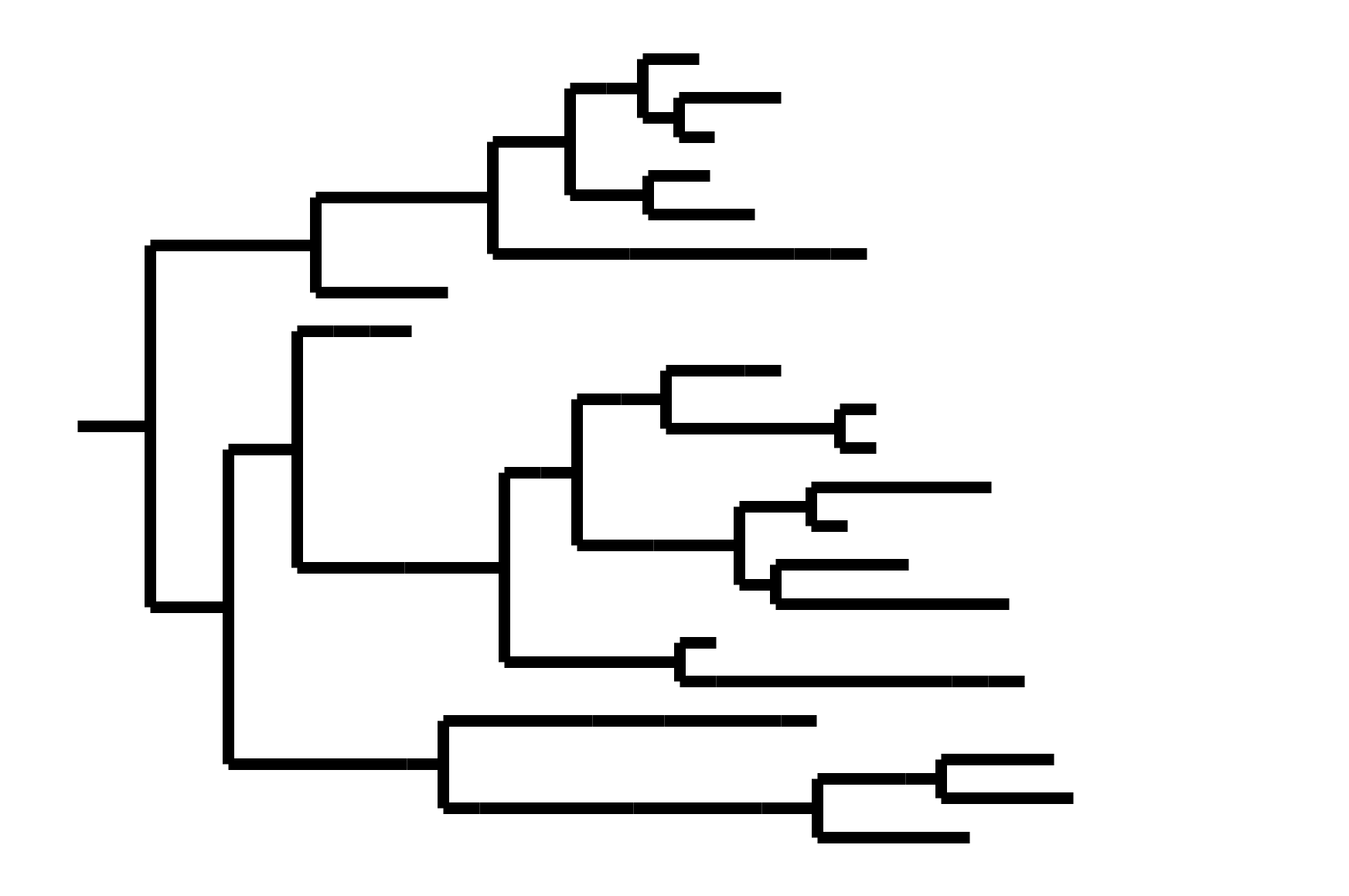
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
CoV-OC43 split into two ladder-like lineages. Influenza B evolves this way too. It's theoretically possible to pick well-matched bivalent vaccine.
CoV-229E has ladder-like tree:
Human influenza A evolves this way too. It's theoretically possible to pick single well-matched vaccine strain.
CoV-OC43 split into two ladder-like lineages. Influenza B evolves this way too. It's theoretically possible to pick well-matched bivalent vaccine.
In non-ladder-like tree, there can be high standing genetic variation. Makes picking vaccine strains difficult.
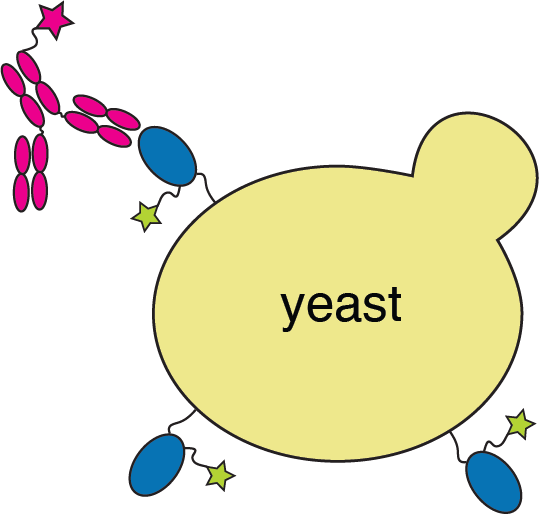
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
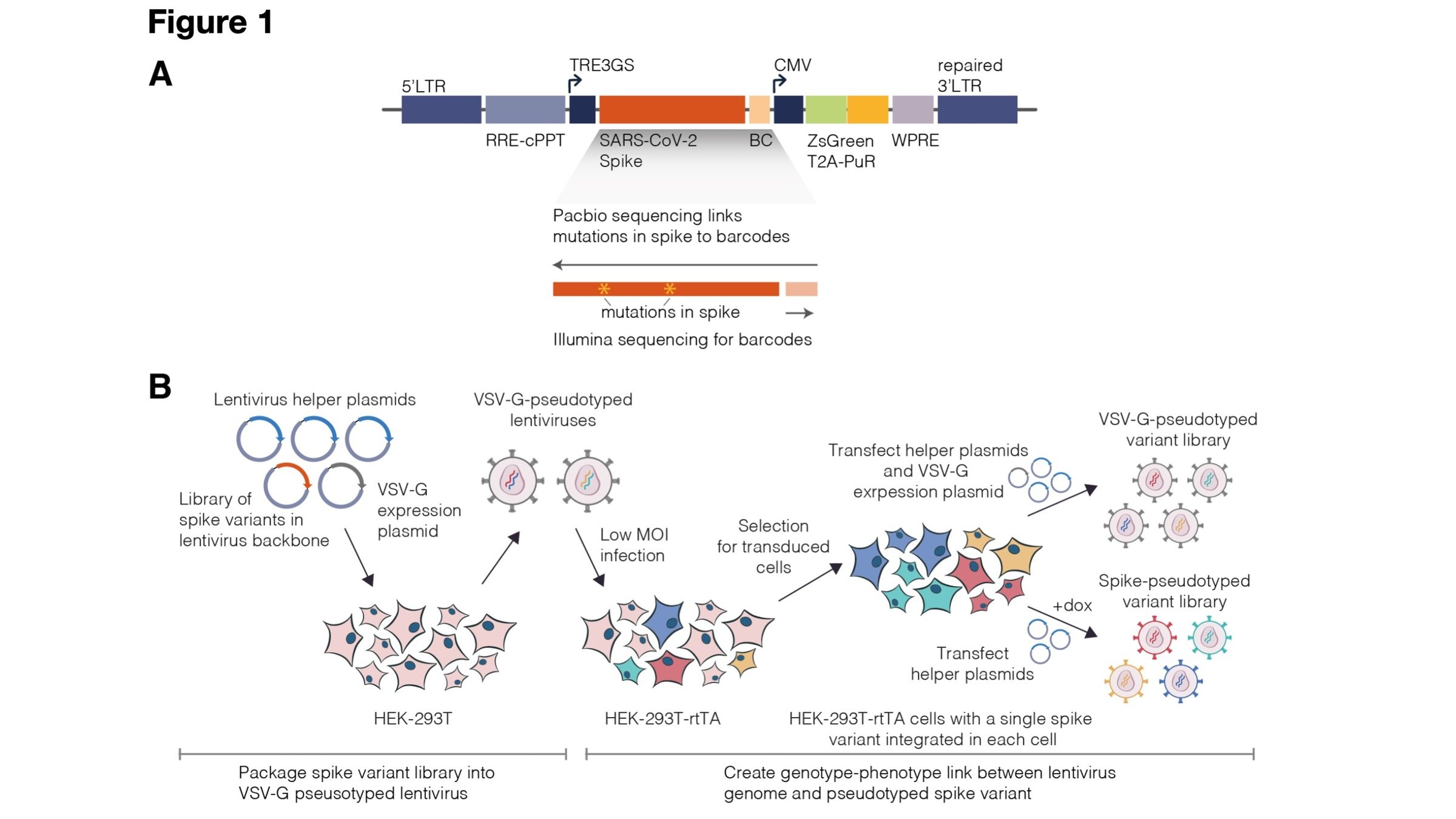
Experiments combine flow cytometry and deep sequencing of a library of yeast expressing all RBD mutants
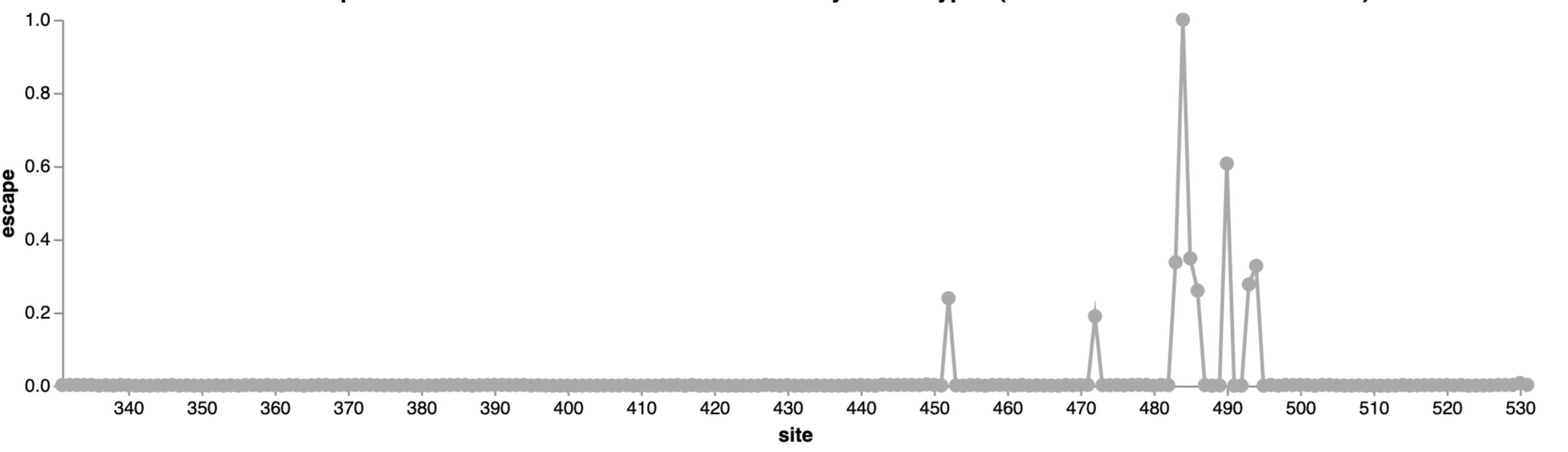
484
452
490
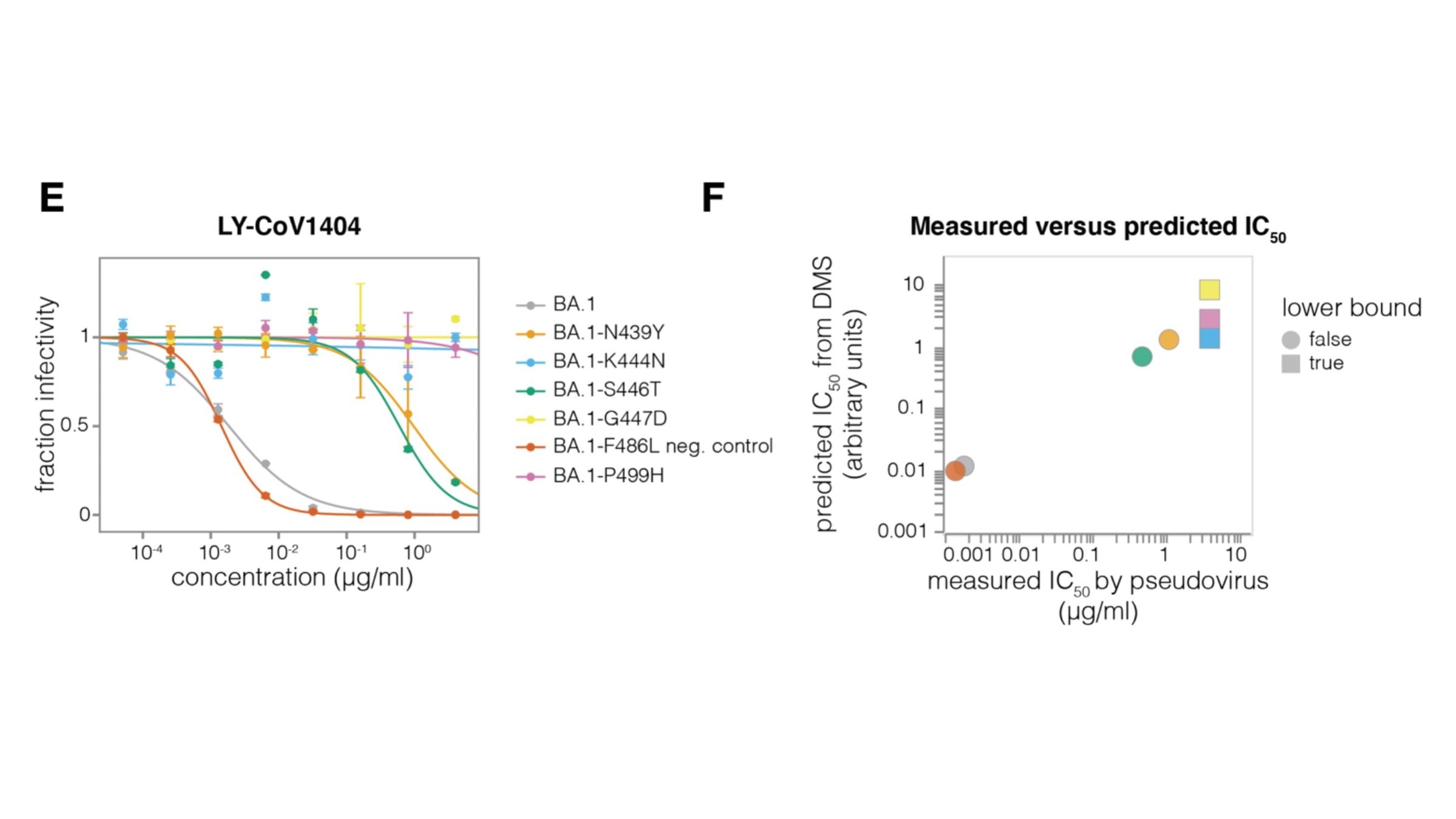
This turned out be an unfortunate choice of an antibody for Eli Lilly to develop as drug, as mutations at sites 484 and 452 were prevalent by early to mid 2021
36 antibodies mapped by Tyler Starr & Allie Greaney in Bloom lab.
36 antibodies mapped by Tyler Starr & Allie Greaney in Bloom lab.
>4,000 (!) antibodies mapped by Yunlong Cao et al at Peking University. See here and here.
Interactive escape calculator: https://jbloomlab.github.io/SARS2-RBD-escape-calc/
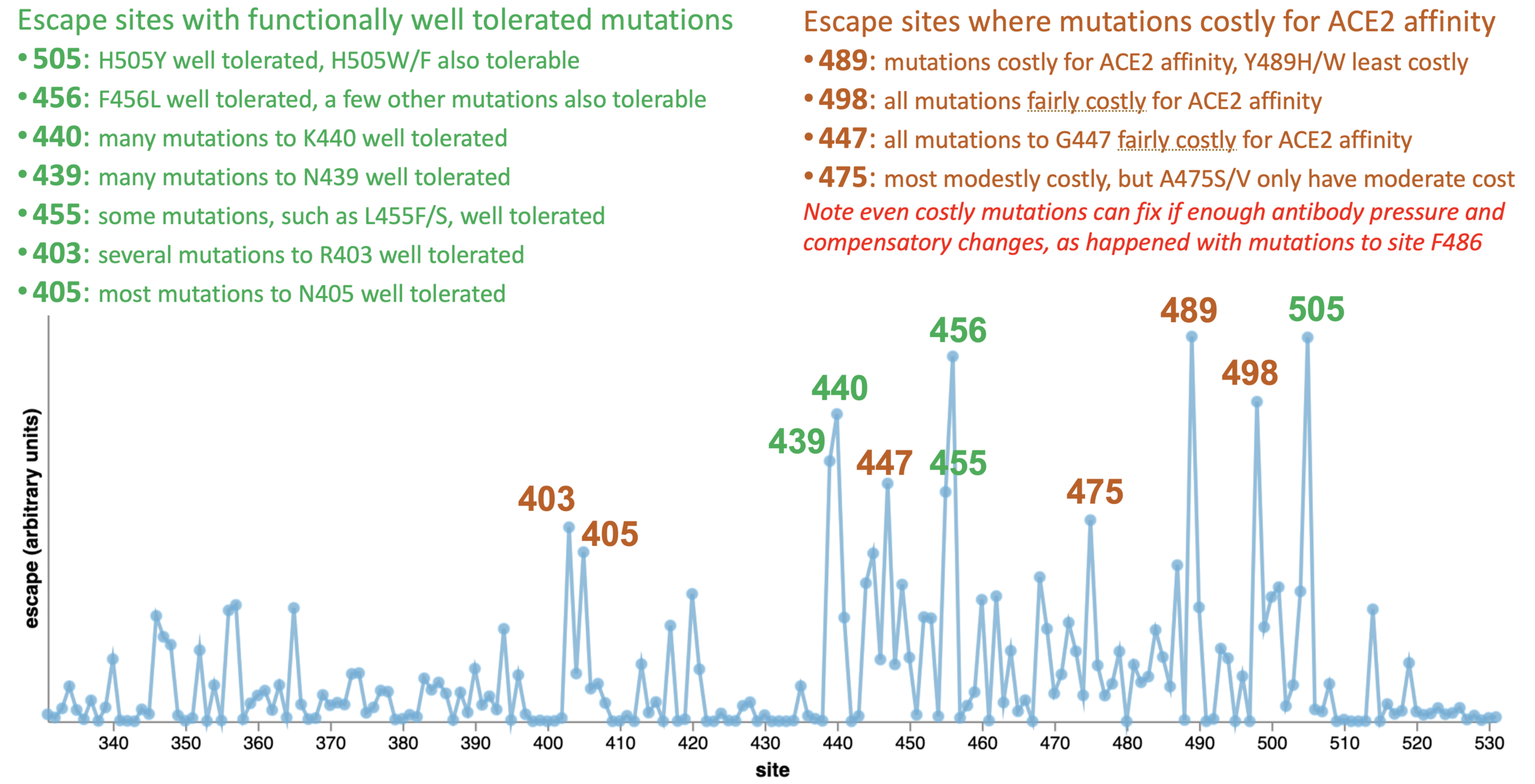
ACE2 affinity from: https://jbloomlab.github.io/SARS-CoV-2-RBD_DMS_Omicron/RBD-heatmaps/
Fitness effects from: https://jbloomlab.github.io/SARS2-mut-fitness/S.html
Bloom lab
Bernadeta Dadonaite
Kate Crawford
Caelan Radford
Tyler Starr
Allie Greaney
Rachel Eguia
rest of lab
University of Washington
Helen Chu and HAARVI cohort
David Veesler
Alex Greninger and UW Lab Medicine
By Jesse Bloom
Interpreting the evolution of SARS-CoV-2



