


Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
I am on the scientific advisory boards of Apriori Bio and Oncorus
I have consulted on topics related to viral evolution for Moderna and Merck
I am an inventor on Fred Hutch licensed patents related to deep mutational scanning of viral proteins
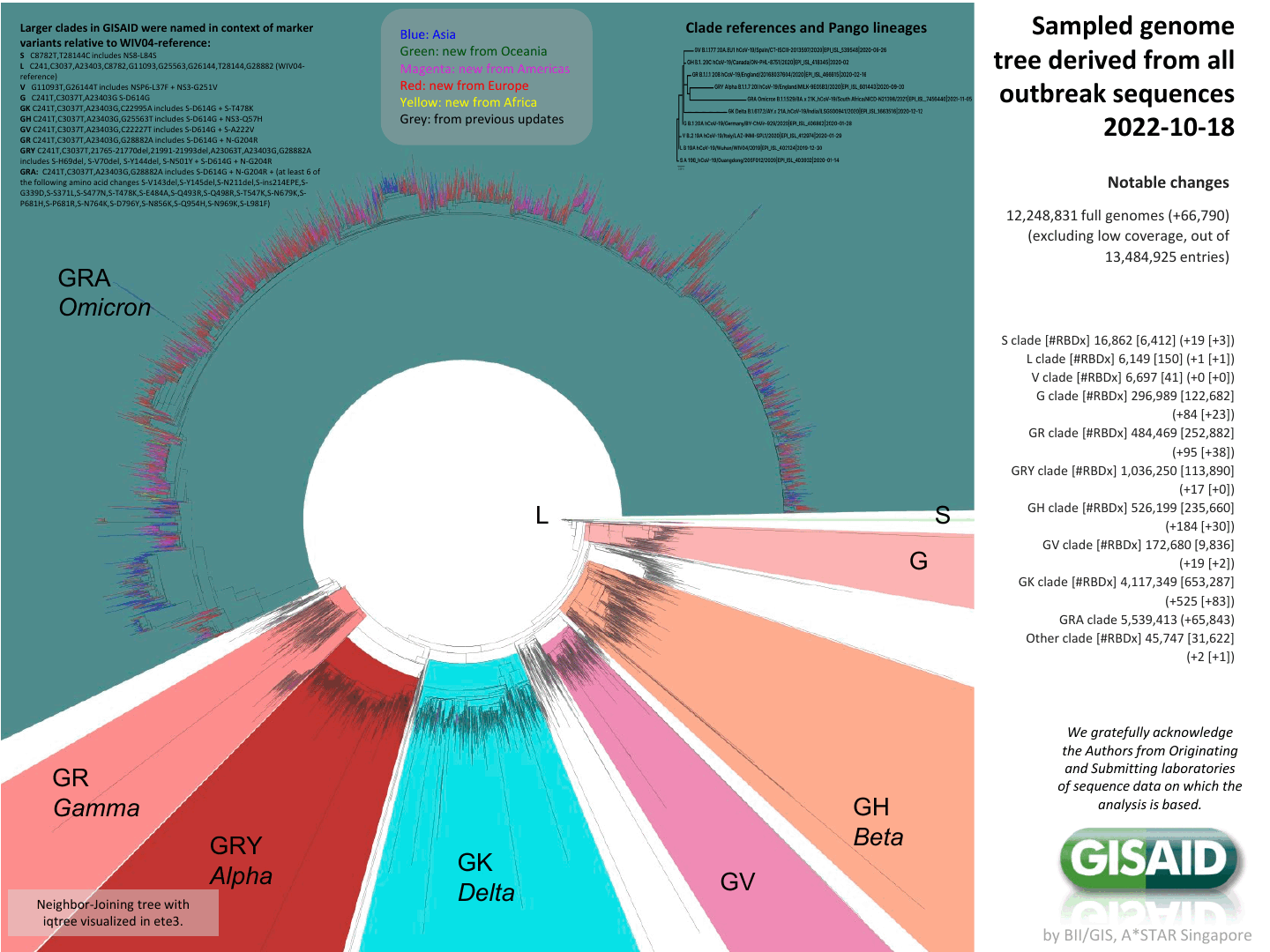
Tree of the ~12-million SARS-CoV-2 sequences in GISAID
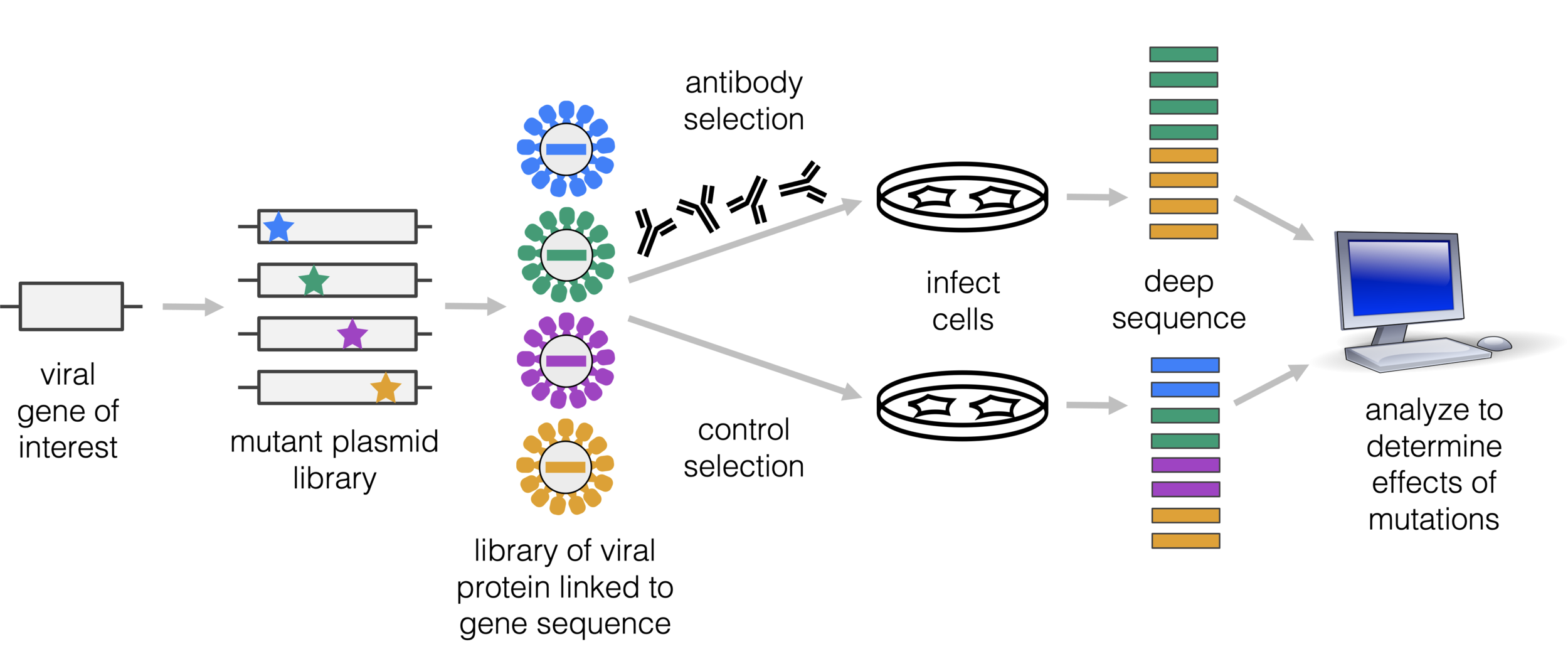
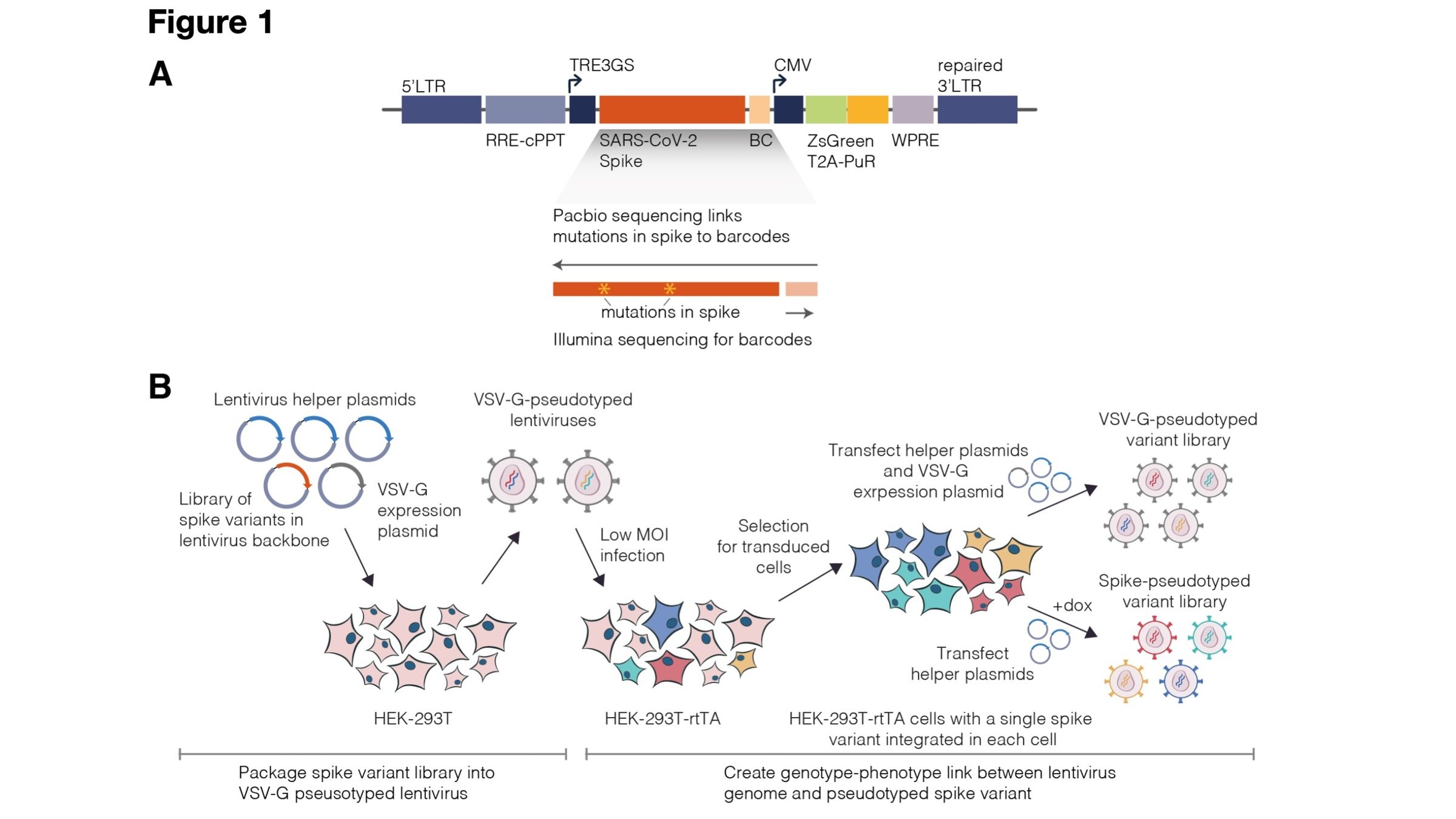
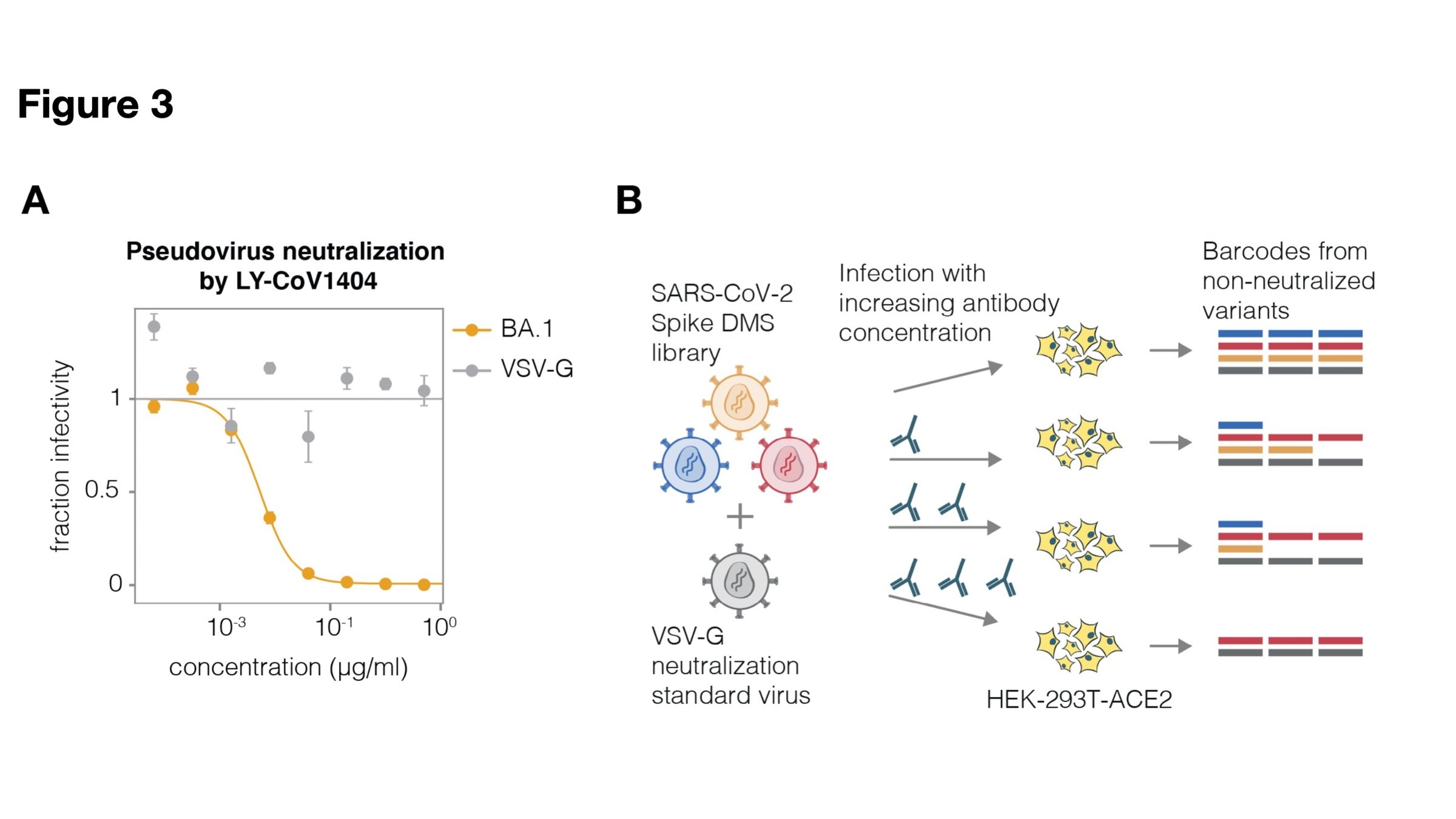
Amenable to multiple ways to link viral protein to gene sequence, and different selections (not just antibodies)
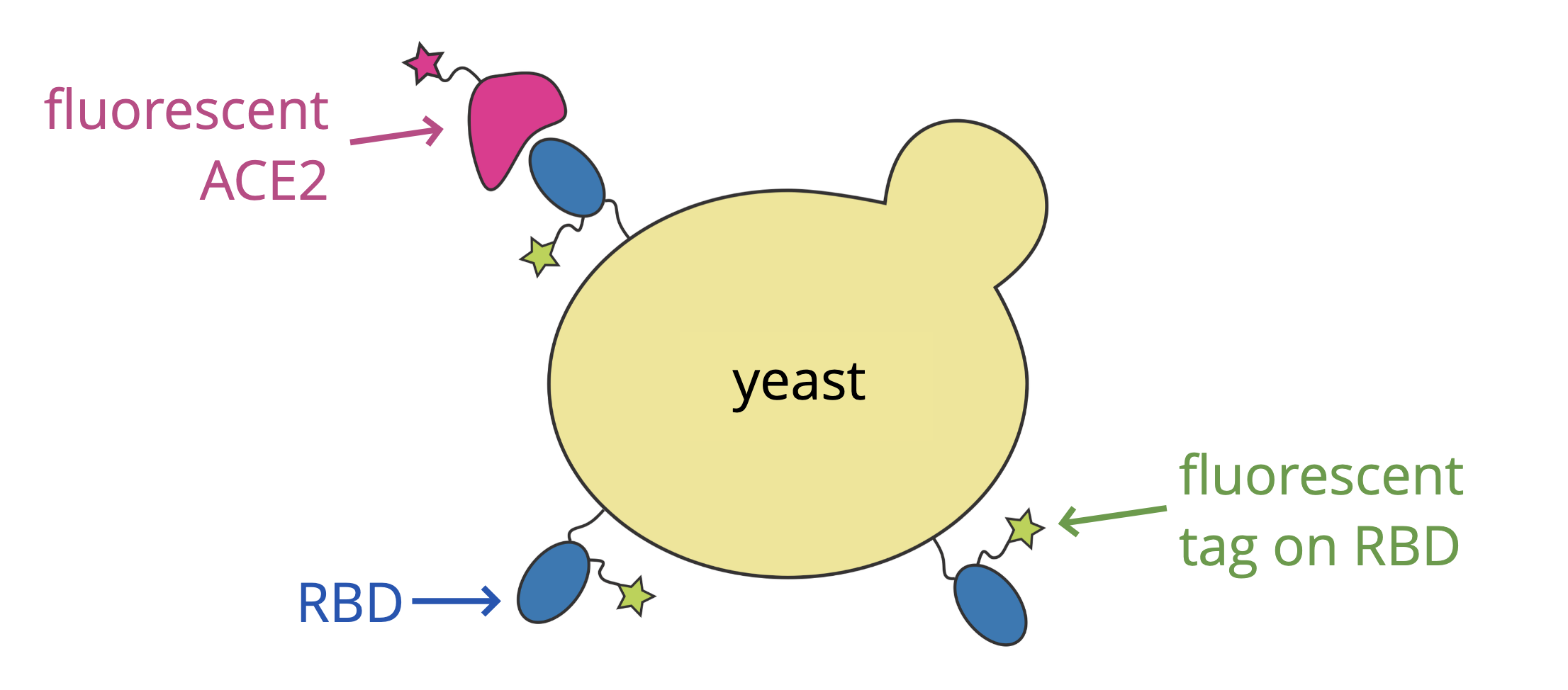
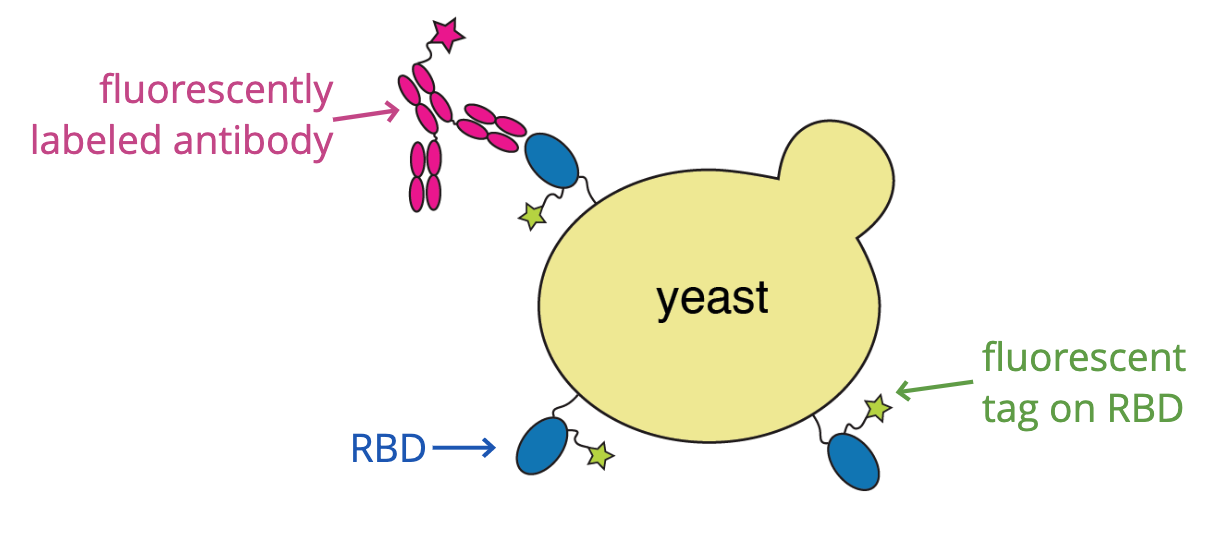
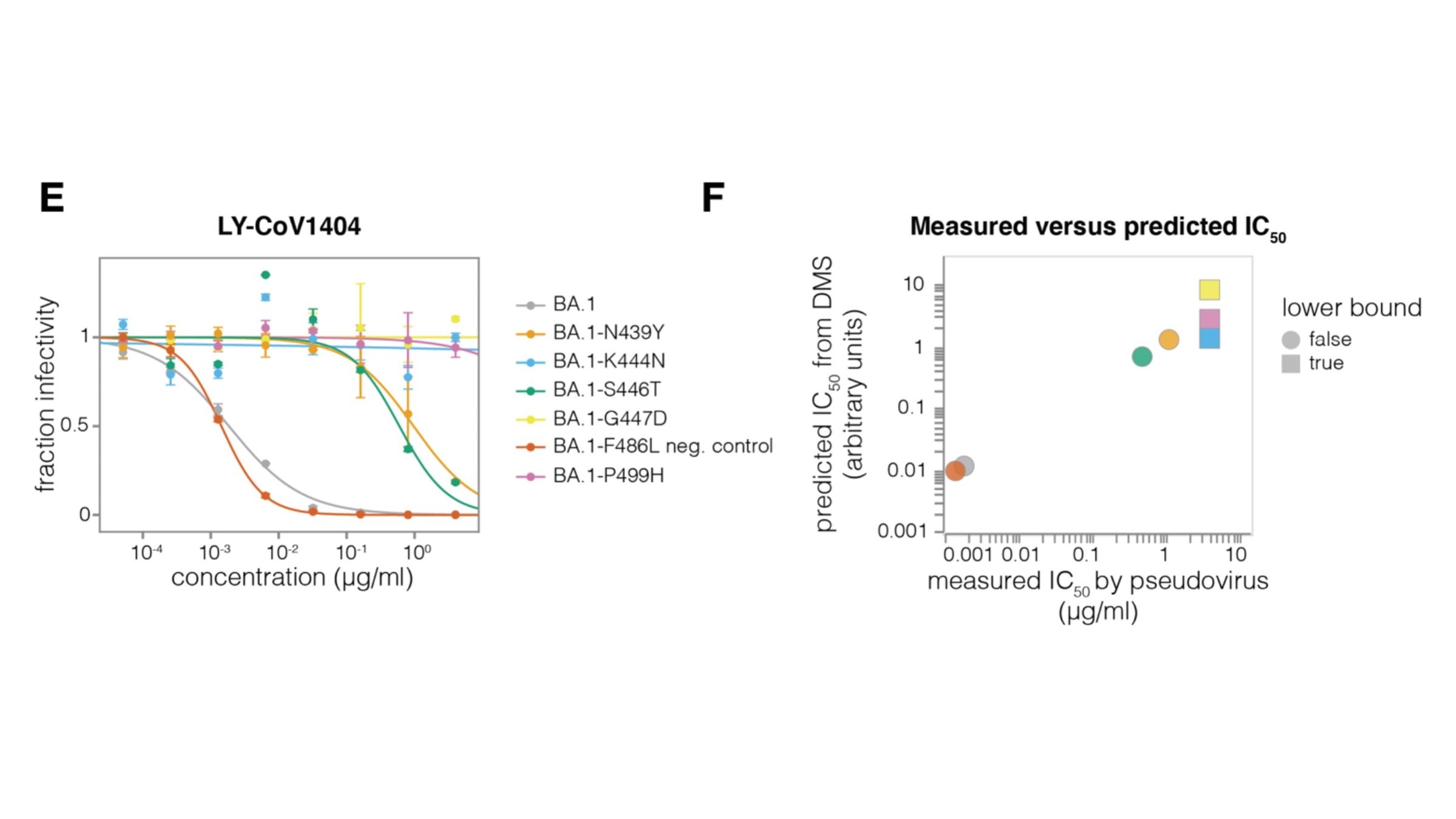
For these studies, we displayed the RBD on the surface of yeast, so we could measure how mutations affects its affinity for ACE2 or ability to escape antibody binding.
Escape calculator is described in Greaney et al (2022), and is available at https://jbloomlab.github.io/SARS2_RBD_Ab_escape_maps/escape-calc/
36 antibodies mapped by Tyler Starr & Allie Greaney in Bloom lab, from early SARS-CoV-2 strains
1,522 (!) antibodies mapped by Sunney Xie, Richard Cao, Fanchong Jian, et al at Peking University. From early strains, BA.1, & patients with prior SARS-CoV-1 infection. See here.
By Jesse Bloom
Viral deep mutational scanning (short version)




