Using association rule mining to determine promising secondary phenotyping hypotheses
BIOINFORMATICS
Anika Oellrich, Julius Jacobsen, Irene Papatheodorou,
The Sanger Mouse Genetics Project and Damian Smedley
U10104025林允中
Genotype(基因型)-Phenotype(表現型)
Phenotype
├Primary Phenotype
└Secondary Phenotype
Gene–phenotype relations are crucial to the improvement of our understanding of human heritable diseases as well as the development of drugs.
However, given that there are ~20000 genes in higher vertebrate genomes and the experimental verification of gene–phenotype relations requires a lot of resources, methods are needed that determine good candidates for testing.
- 了解人類遺傳疾病並開發藥物
- 基因數量龐大→利用實驗來驗證基因型-表型將耗費許多資源
動機
We hypothesized that phenotypes that significantly co-occur with each other more often than expected by chance, given the overall amount of phenotype annotations, constitute good candidates for the secondary phenotype experiments.
For example, it is known that body weight correlates with bone density or grip strength and changes in body weight often lead to changes in the correlated phenotypes.
Using a large dataset of phenotype annotations, we can determine pairs of phenotypes that may be biologically linked.
- 假定 1.表型共同發生的情形,比預期還要常出現 2.大量的資料可以產生較好的結果
- 如:體重可能與骨質密度或抓握力有關
- 使用大量的表型資料,可以發掘哪些表型在生物學上可能是互相關聯的
方法
流程
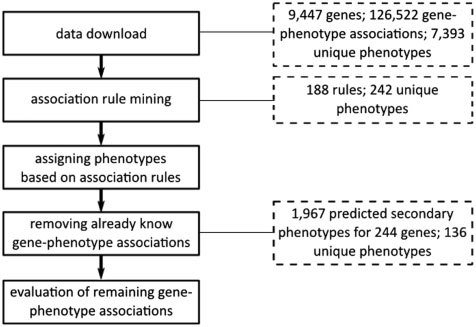
流程
We used the apriori software implementation with the following parameter settings:
-tr -s-6 -m2 -n2 -c90 -ep -v ‘‘%e’’
-tr: to enforce the output of association rules instead of item sets
-s-6: to obtain only rules that are supported by at least six item sets
-m2: to include only rules with a minimum of two items
-n2: to include only rules with a maximum of two items
-c90: to only allow rules with a confidence of 90%
-ep: to provide P-values for each rule
-v “%e”: to add the P-value separated by space to each of the rules.
流程
Rules are returned together with their corresponding P-value to enable potential further filtering and user confidence, e.g.
MP:0004725<- MP:0009448 0
MP:0005606<- MP:0009448 3.9905e-212
MP:0005606<- MP:0009557 4.22726e-182
MP:0000245<- MP:0011171 2.64518e-125
評估
We assume that the secondary phenotype predictions once added to the annotations assigned in the primary screens improve consistency and completeness of the phenotype data.
假定若將secondary phenotype的預測資料加入primary的篩選中,將會改善表型資料的一致性及完整性
To assess the biological coherence without and with the predicted secondary phenotypes, we generated gene clusters based on the primary phenotypes solely, as well as clusters based on the primary and secondary phenotype data in conjunction.
為了評估這個假說,我們產生了兩個資料群,一個全為primary phenotype,而另一組則為primary phenotype和secondary phenotype交集
評估
To assess the performance of a ranking algorithm, commonly Receiver Operating Characteristic (ROC) curves are used that are calculated based on a benchmark dataset. In our case, we used known gene–disease associations to assess the value of the predictions. If the secondary phenotypes add value to the predictions, then a performance increase should be visible from the Area Under Curve (AUC) of the two ROC curves (one for the predictions based on primary phenotype data, and one for the predictions based on primary and secondary phenotype data). To test whether the increase in the AUC is significant, we used a two-sided test for ROC curves available online.
結果
Using a two-sided Wilcoxon signed-rank test (as implemented in R, α=0.05), we obtained a P-value of 2.2x10^-16, indicating a significant improvement when adding secondary phenotype annotations to the previously confirmed in the primary phenotype scans.
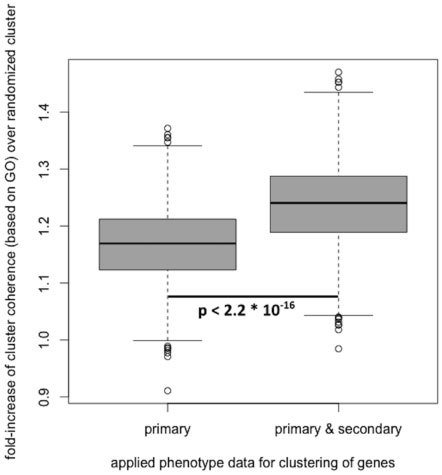
These results suggest that the predicted secondary phenotypes possess biological validity but will have to be experimentally verified in secondary phenotype screens.
Predicted secondary phenotypes significantly improve the biological coherence of gene clusters
結果
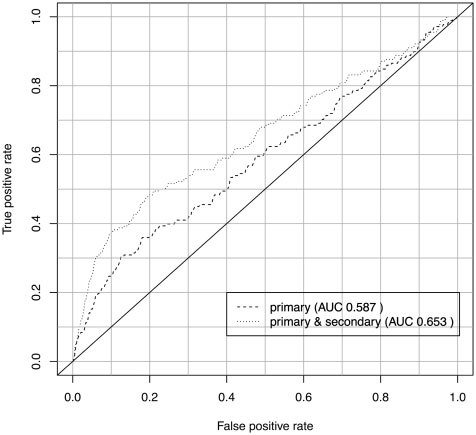
In addition to assessing the biological coherence of gene clusters, we also verified the predicted secondary phenotype annotations by applying them in a second application use case: the prediction of disease gene candidates.
For both predictions, we calculated the ROC curves, using gene–disease associations contained in MGD as benchmark dataset. Both the obtained ROC curves are depicted in Figure 4. Applying a onetailed Student’s t-test (α=0.05) to the ROC curves, we obtain a P-value of P=0.02.
Secondary phenotypes significantly improve the
predictability of disease gene candidates
結論
- Predicted secondary phenotypes improve the biological coherence of the clusters.
- Secondary phenotypes improve the predictability of causative disease genes.
- Secondary phenotypes further characterize genes assessed with primary phenotyping.
Using association rule mining to determine promising secondary phenotyping hypotheses
By lzong



