



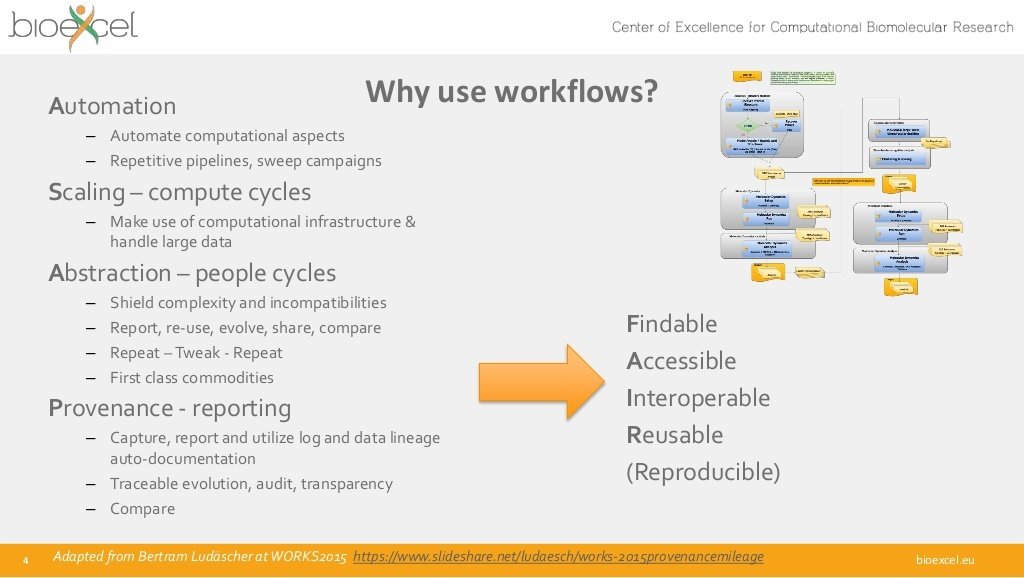
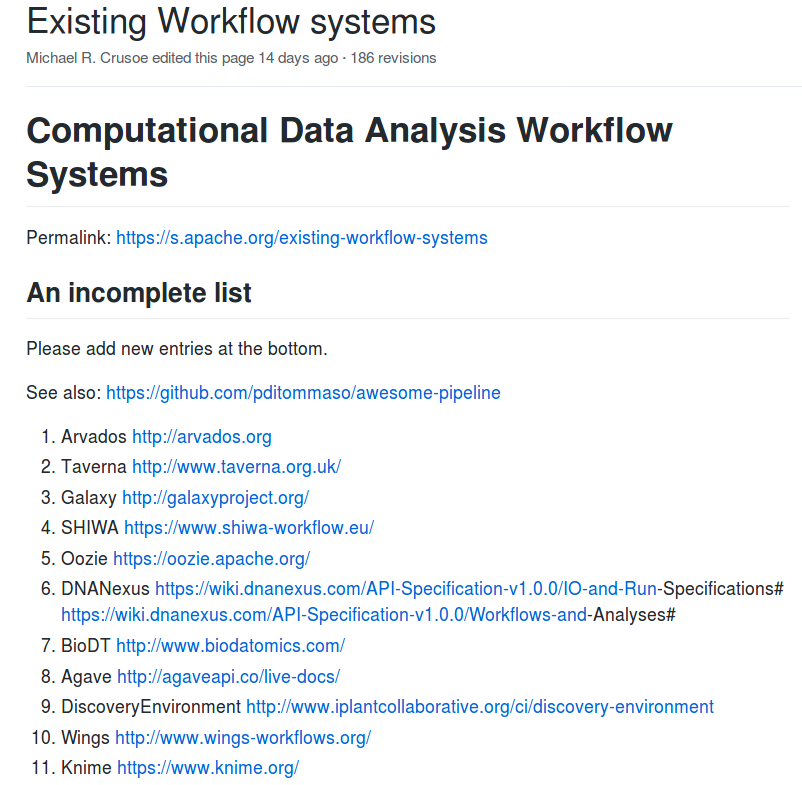
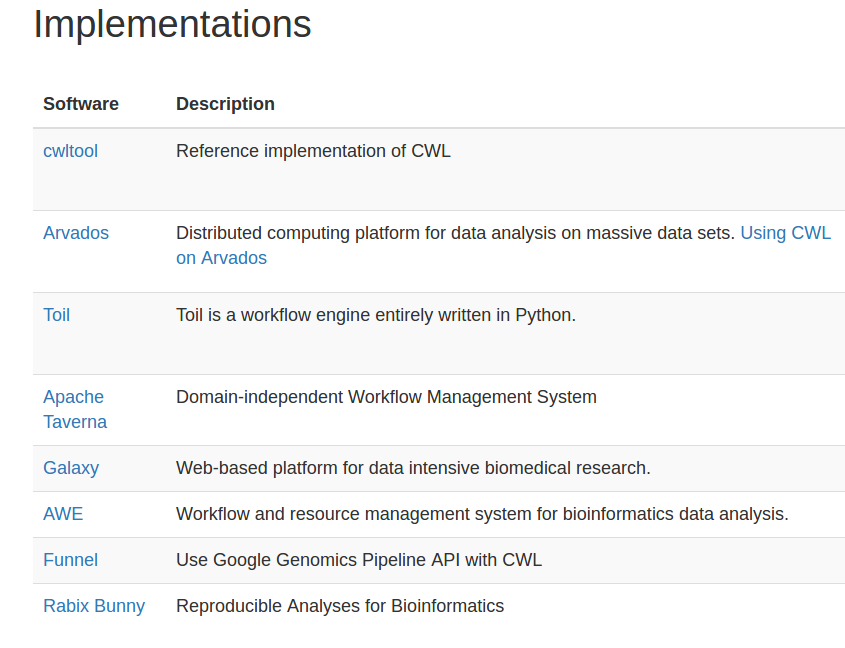
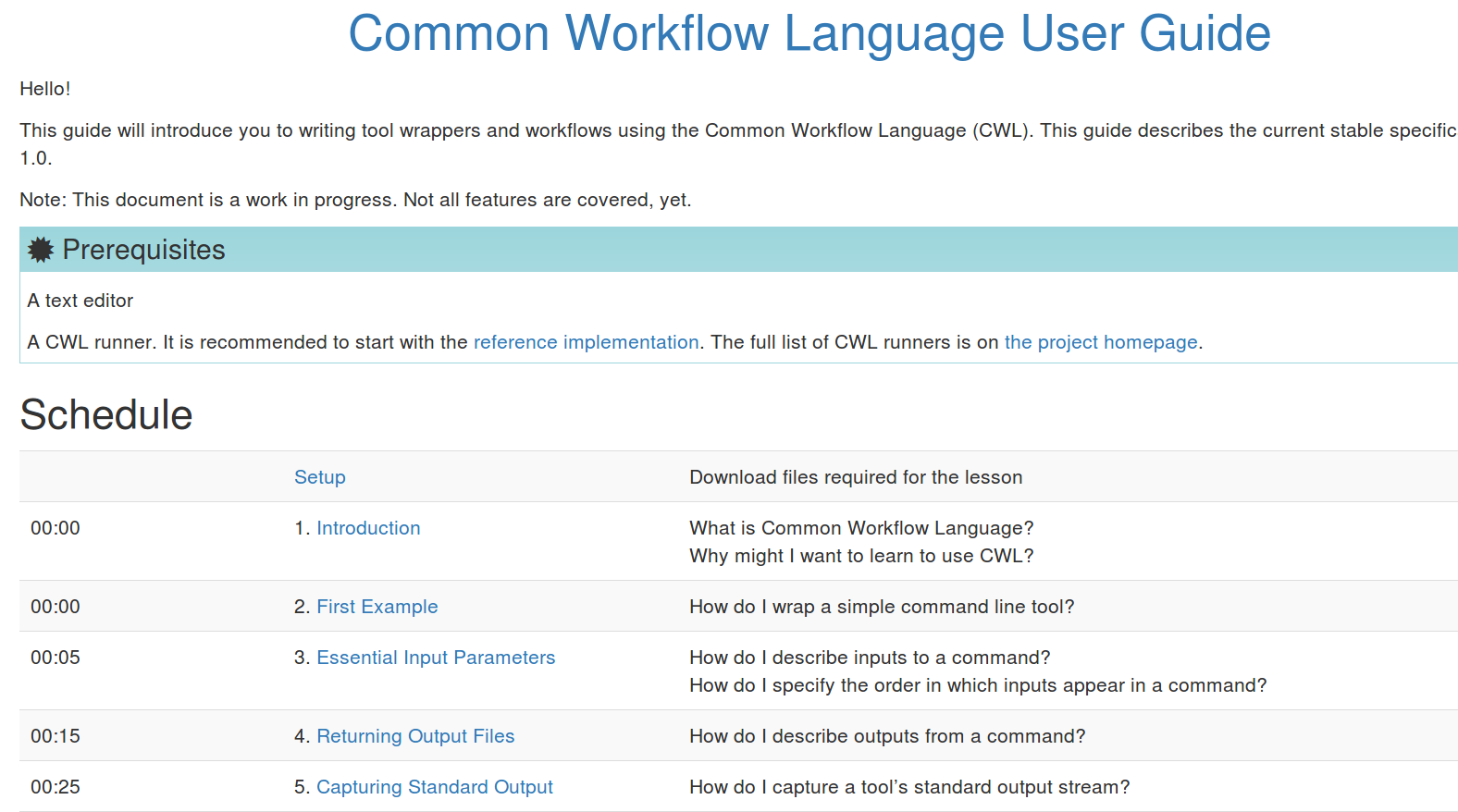


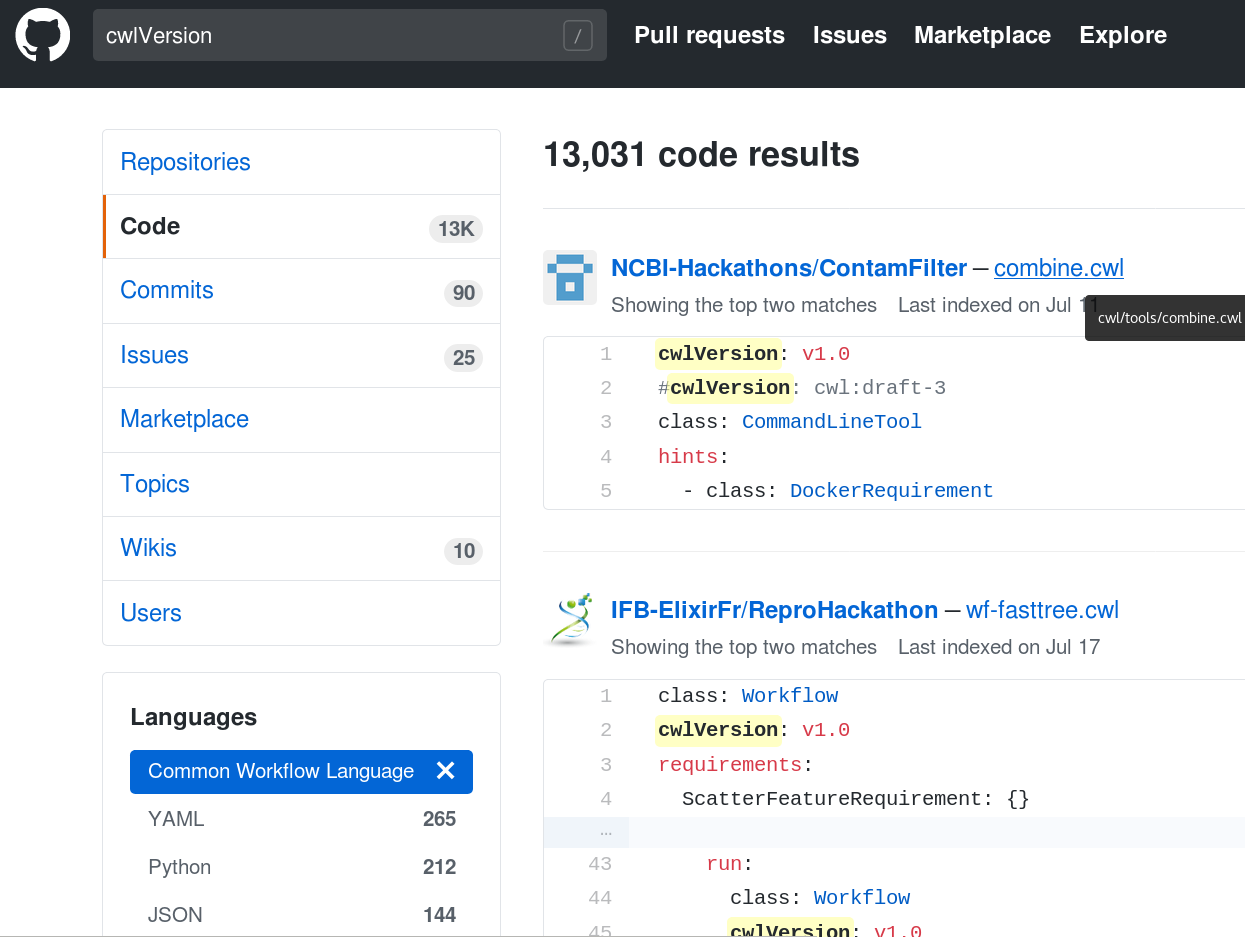











Stian Soiland-Reyes PRO
Senior Lecturer at eScience Lab, Dept of Computer Science, The University of Manchester. Open Source research software engineer. Interest: Linked Data, Web, provenance, annotations, Open Science, reproducible research results.




