Postdoc Interview Roche
Mechthild Lütge
04.12.2023
2016
Joined Bork group at the EMBL Heidelberg as a Masters student
Bacterial pan-genomes:
- functional annotation
- co-occurrence analysis
- selective pressure analysis
2018
Joined Immunobiology group at the Kantonsspital St.Gallen as a PhD student
Single cell transcriptomics:
- setup pipeline for automated mapping/pre-processing
- establish routines for independent data exploration and standardize analyses
Immune cell niches
- Stromal cell niches across murine lymphoid organs
- Human lymph node stromal cells
Bacterial genomic data from large database freeze
Research experience in applied bioinformatics
>30 projects:
scRNAseq (10X, smartSeq), scVDJ, spatial transcriptomics (Visium), microbiome data (16S), metagenome data,
Mapping pipeline
Idea:
- reduce workload
- standardize processing to secure comparability and reproducibility
seurat.rds

Download rawdata
Mapping to reference genome
Quality control and filtering
Normalization, dimensionality
reduction, clustering
Data exploration
Makefile:
- wget
- cellranger
- R script
### sample specifications as specified in config.json
## runName := $(shell python3 config.py runName) ...
### exclude all and clean as targets
.PHONY: all clean debug
### require all sample names to be processed
all: $(sampleName)
debug:
@echo "runName: $(runName)"
@echo "sampleName: $(sampleName)"
@echo "projectPath: $(projectPath)"
@echo "referenceFasta: $(referenceFasta)"
@echo "referenceGtf: $(referenceGtf)"
@echo "referenceDir: $(referenceDir)"
@echo "organism: $(organism)"
@echo "sceScript: $(sceScript)"
$(sampleName): /data/raw/$(runName)/$(sampleName).tar $(referenceDir)/reference.json /data/processed/$(runName)/$(sampleName)_seurat.rds
@echo "Processing Sample: $@"
### Download sample
/data/raw/$(runName)/$(sampleName).tar:
@echo "Downloading Sample: $(sampleName)"
wget -r -nH --cut-dirs=2 --no-parent --reject="index.html*" -e robots=off --user $(user) --password $(password) $(baseURL)$(runName)/$(sampleName).tar \
--directory-prefix /data/raw/
### Create reference
$(referenceDir)/reference.json:
@echo "Creating Reference: $@"
cd /data/reference && $(cellrangerPath)/cellranger mkref --nthreads=32 --genome=$(notdir $(referenceDir)) --fasta=$(notdir $(referenceFasta)) \
--genes=$(notdir $(referenceGtf))
### Extract, map and process sample
/data/processed/$(runName)/$(sampleName)_seurat.rds: /data/raw/$(runName)/$(sampleName).tar
@echo "Extracting Sample: $(sampleName)"
mkdir -p /data/tmp/$(runName)
tar -xf /data/raw/$(runName)/$(sampleName).tar --directory /data/tmp/$(runName)
@echo "Mapping Sample: $(sampleName)"
mkdir -p /data/mapped/$(runName)
cd /data/mapped/$(runName) && $(cellrangerPath)/cellranger count --id=$(sampleName) --fastqs=/data/tmp/$(runName)/$(sampleName) \
--sample=$(sampleName) --nosecondary --transcriptome=$(referenceDir) --localcores=32
rm -rf /data/tmp/$(runName)/$(sampleName)
@echo "Processing Sample: $(sampleName)"
R CMD BATCH "--args /data/mapped/$(runName)/$(sampleName)/outs/filtered_feature_bc_matrix $(organism) $(runName) $(sampleName) \
/data/processed/$(runName)/$(sampleName)_seurat.rds" $(sceScript)
### Clean up generated data
clean:
rm -rf /data/raw/$(runName) /data/tmp/$(runName) /data/mapped/$(runName) /data/processed/$(runName)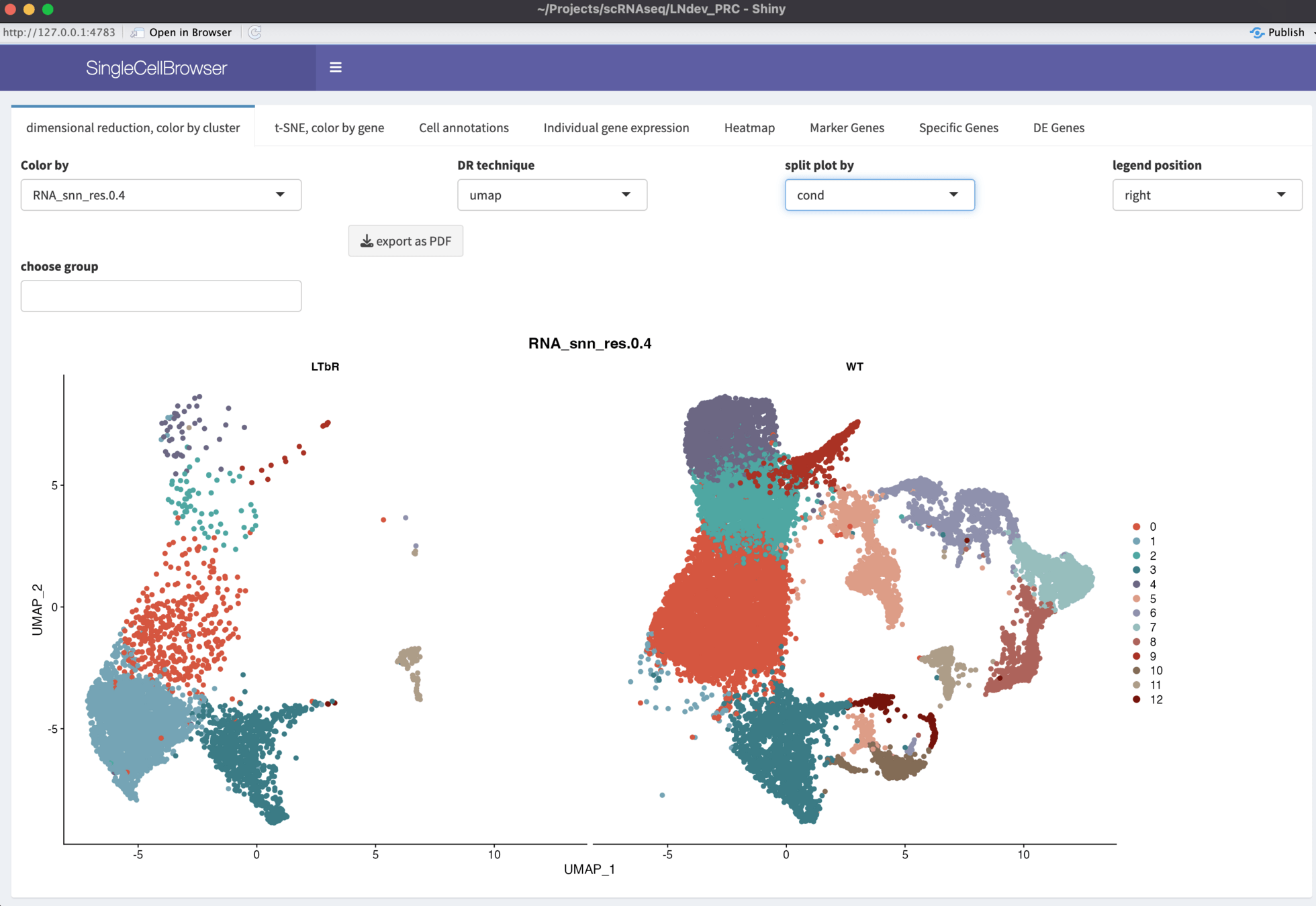
Mapping pipeline
Idea:
- reduce workload
- standardize processing to secure comparability and reproducibility
Cron Job:
0 22 *** (every 24 hours)
Download rawdata
Mapping to reference genome
Quality control and filtering
Normalization, dimensionality
reduction, clustering
Data exploration
Makefile:
- wget
- cellranger
- R script
seurat.rds
pipeline.py:
- create config.json
- make
fetch_sample.bash:
- check for new samples
- python pipeline.py


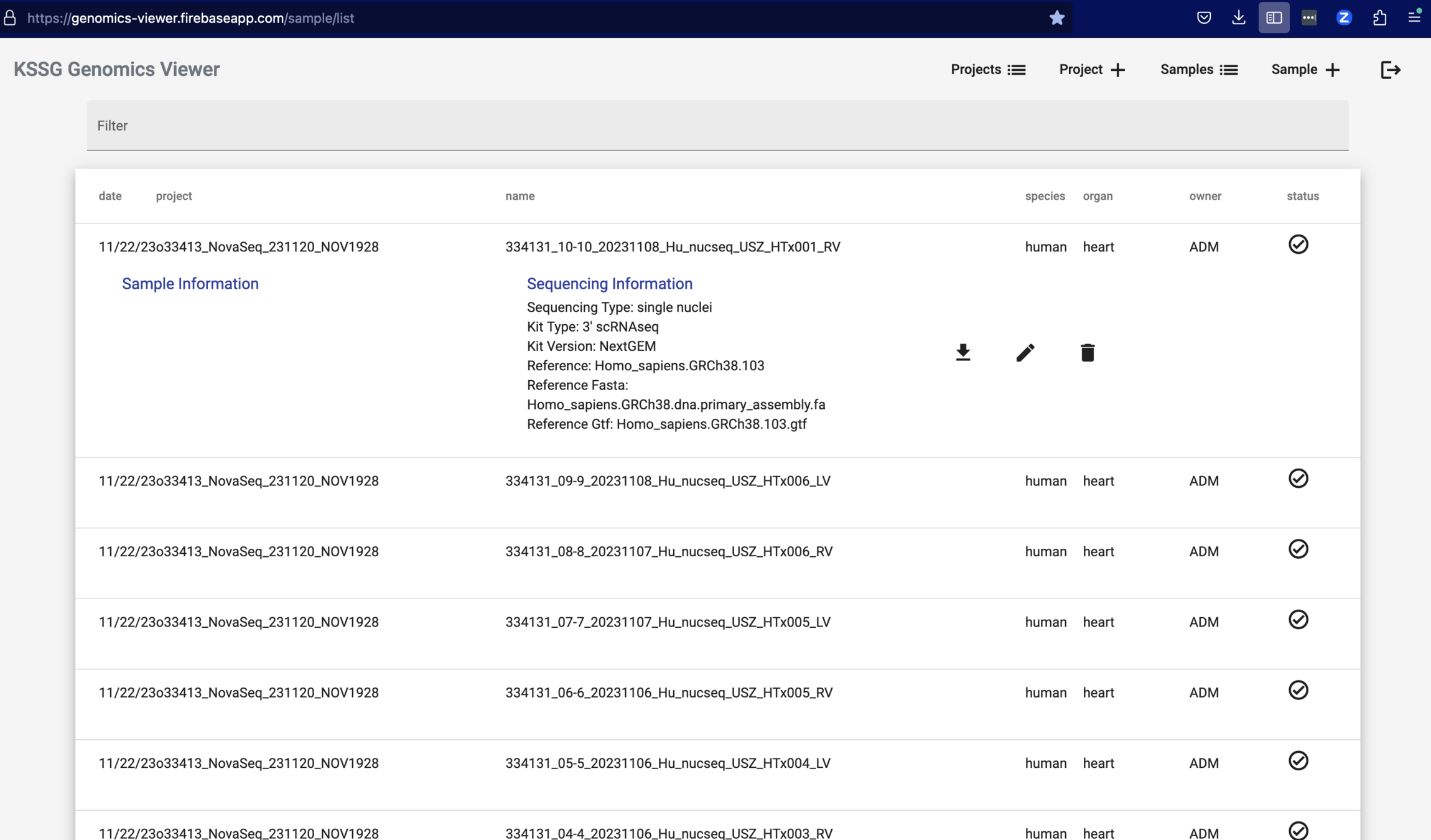
Genomics Viewer:
Web App to access and edit database with all samples
Downstream analyses - How to decide for a tool?
Considerations:
- What is my biological question? What do I want the tool to do?
- On what input data depends the analysis?
- How can I evaluate the output?
Project idea:
Localization of perivascular reticular cells in human lymph nodes using spatial transcriptomics and scRNAseq data → tool for celltype decomposition
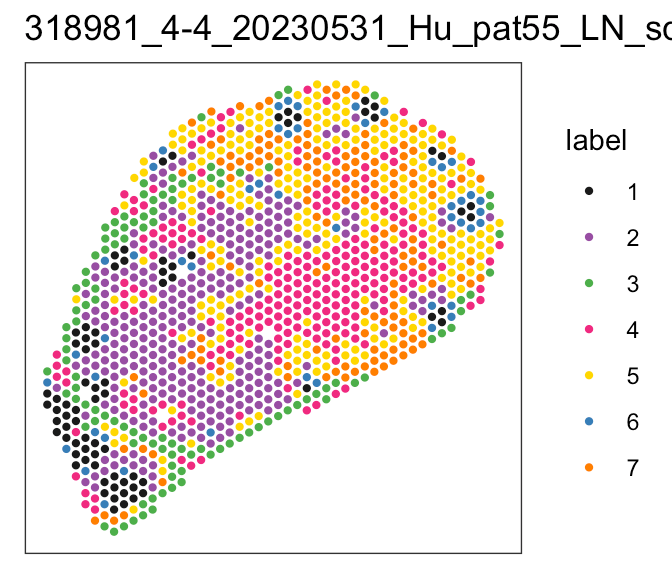
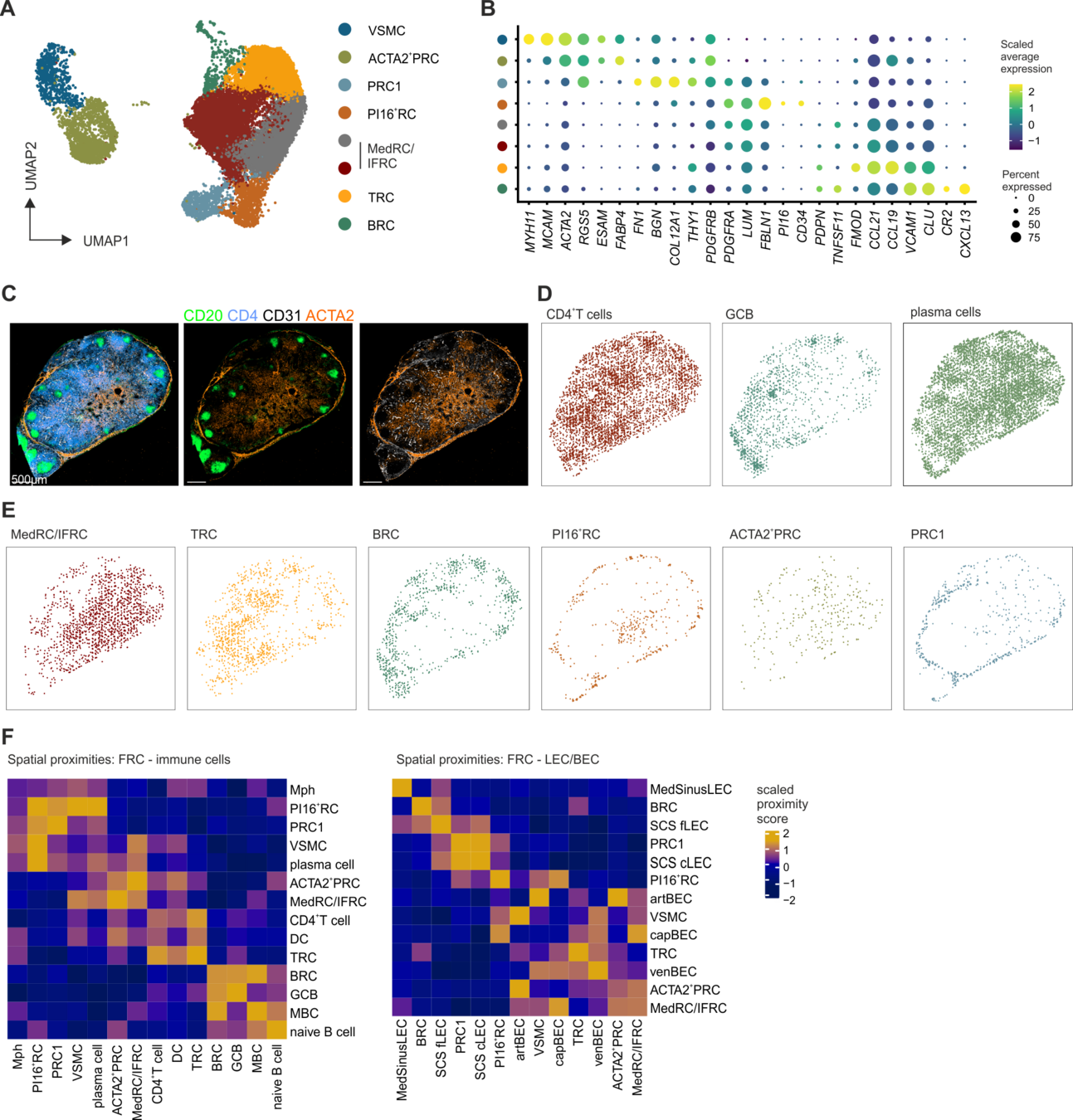
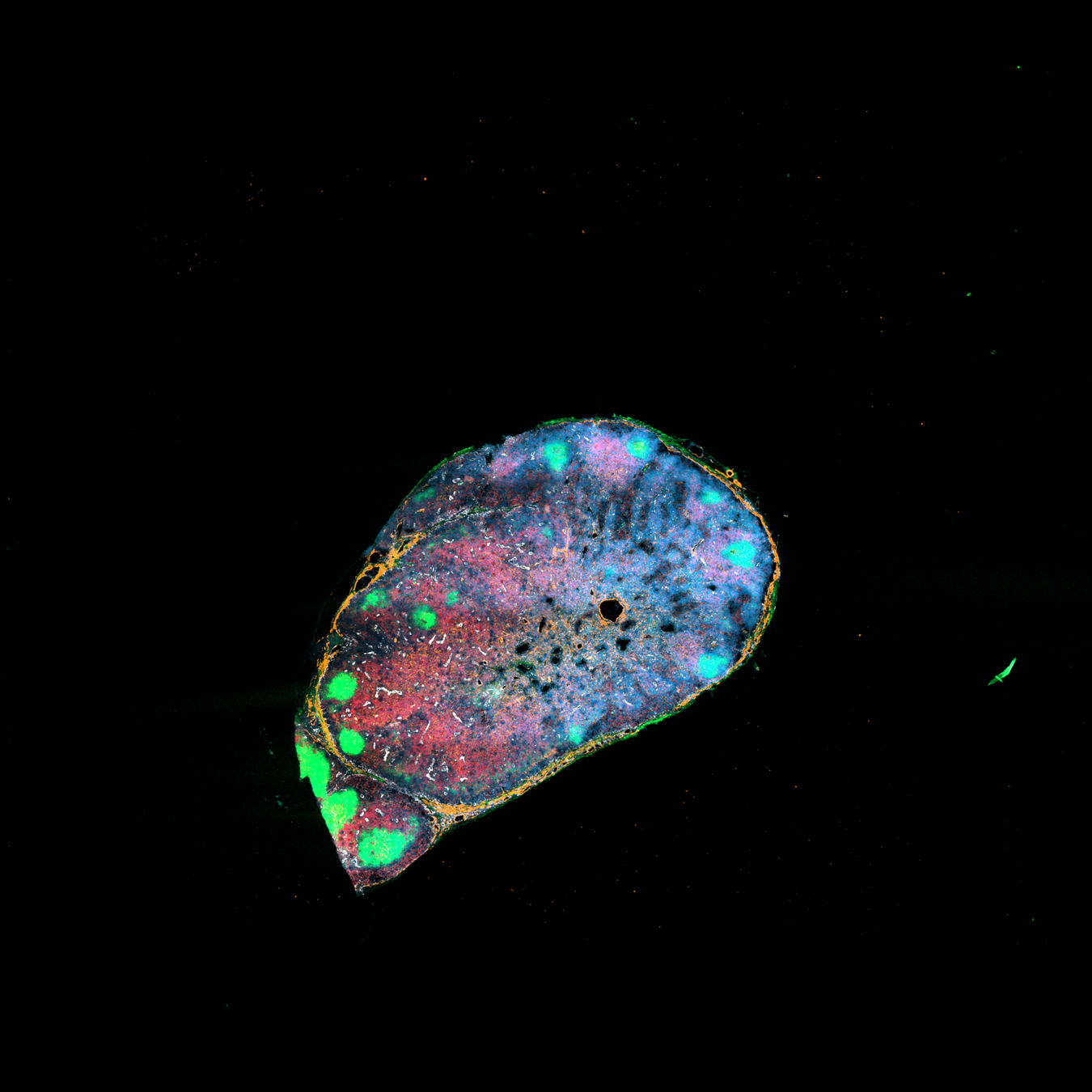
Perivascular reticular cells
Histological images, biological knowledge about lymph node architecture
Spatial transcriptomics + single cell reference
Select tools to test:
- Literature (benchmarks?), Recommendations
- Feasibility (implementation, computing time, ...)
SpaTalk, RCTD (spacexr)
Downstream analyses - How to decide for a tool?
SpaTalk:
Decomposition based on non-negative linear model
RCTD:
Maximum-likelihood estimation to resolve a statistical model that estimates mixtures of cell types at each pixel assuming gene counts to be Poisson distributed
Downstream analyses - How to decide for a tool?
Define evaluation criteria:
- B cell, T cell, endothelial cell position
- Expression of marker genes
- Known fibroblasts compartments
- Celltype proportions from FACS data
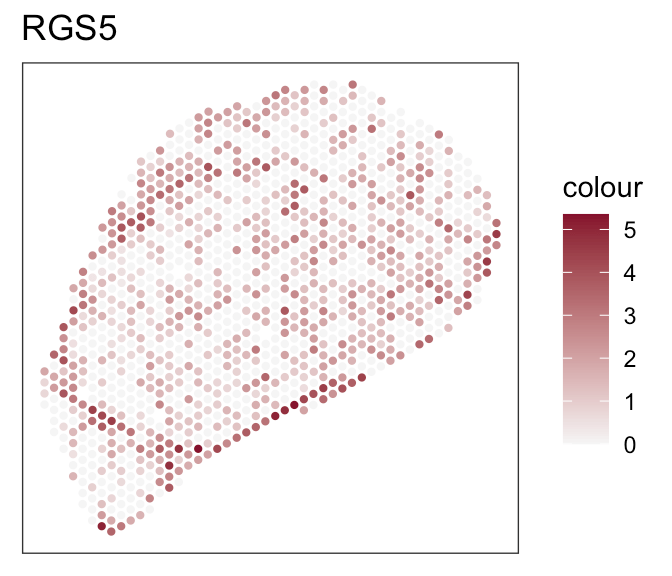
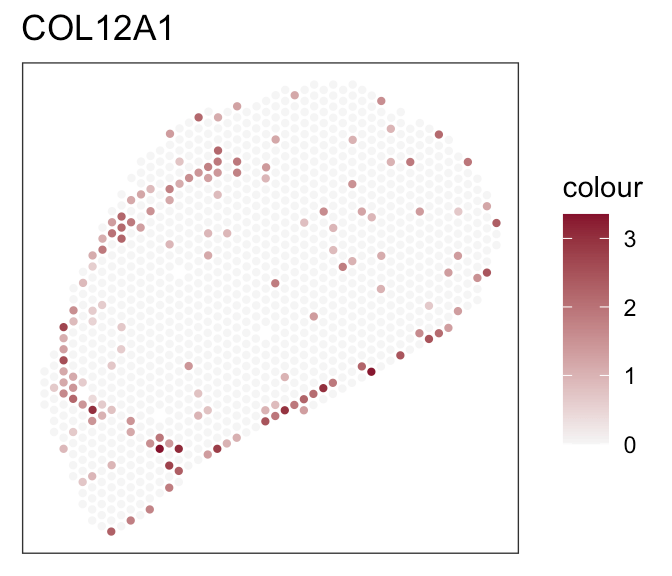
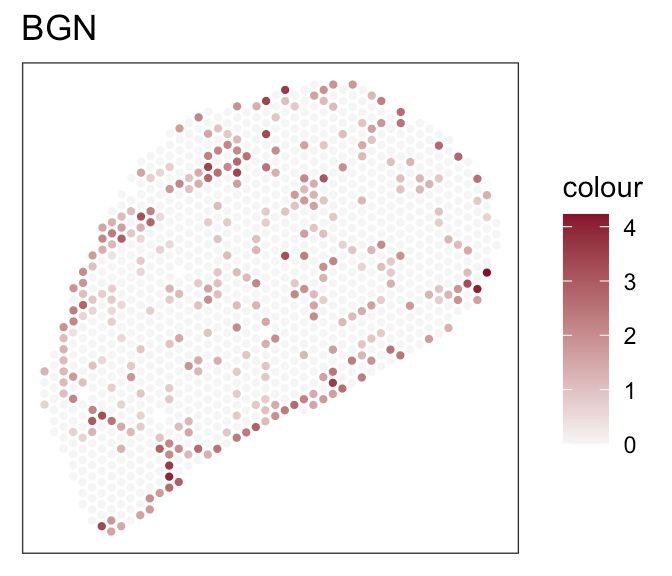
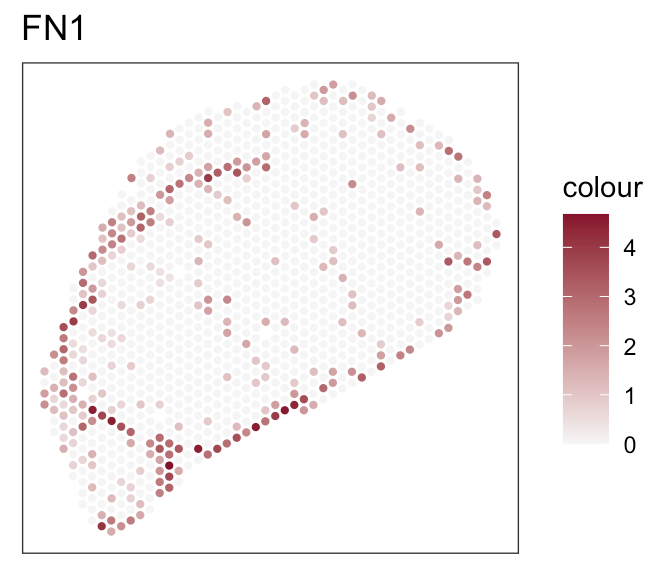
Downstream analyses - How to decide for a tool?
Iterative testing and parameter optimization for each tool:
- Run tool with default parameters
- Evaluate output based on evaluation criteria
- Adjust parameters/ test different parameter setting
- Re-run
- Both tools achieve robust and comparable outputs
- SpaTalk closer in resembelling celltype proportions
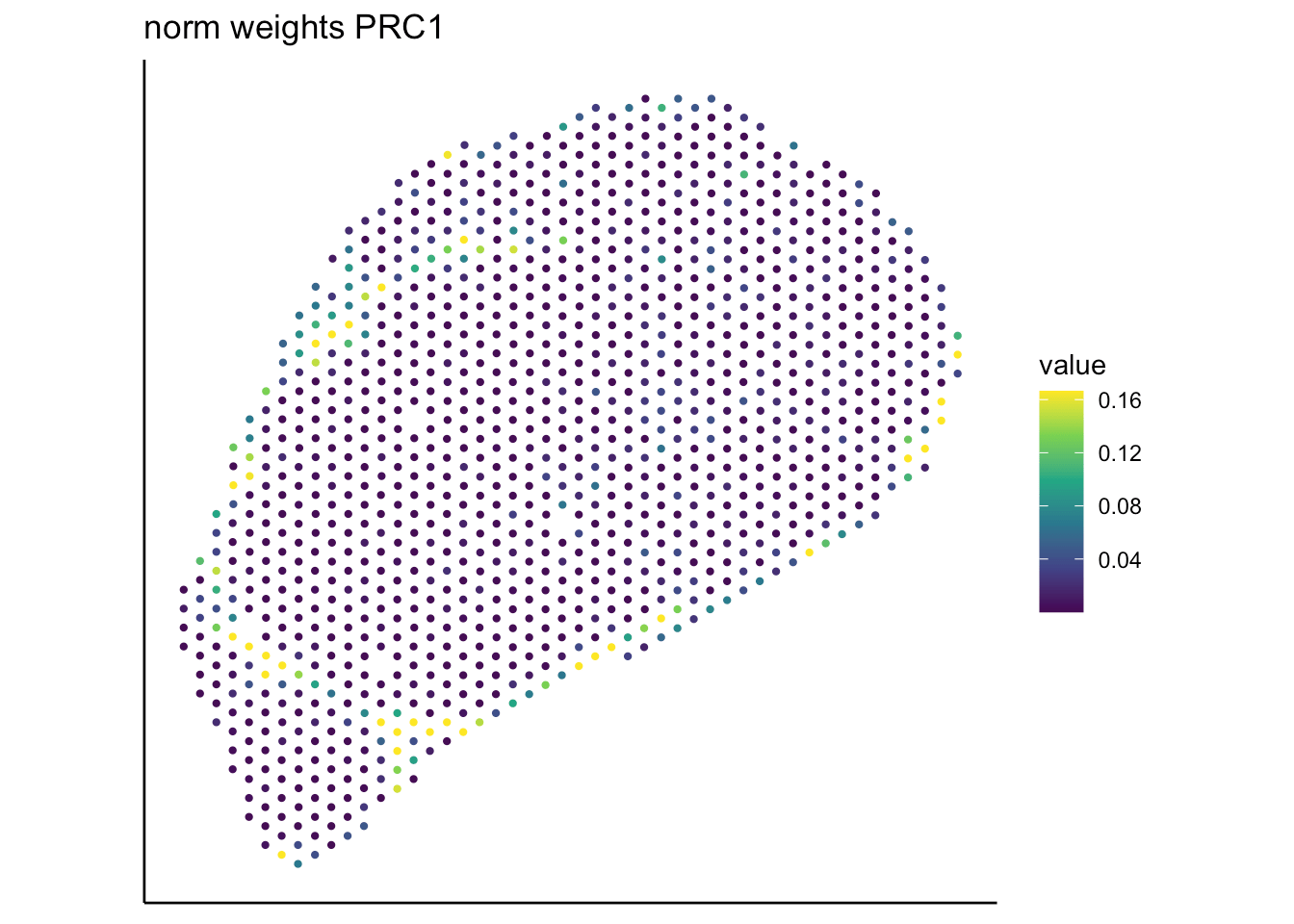
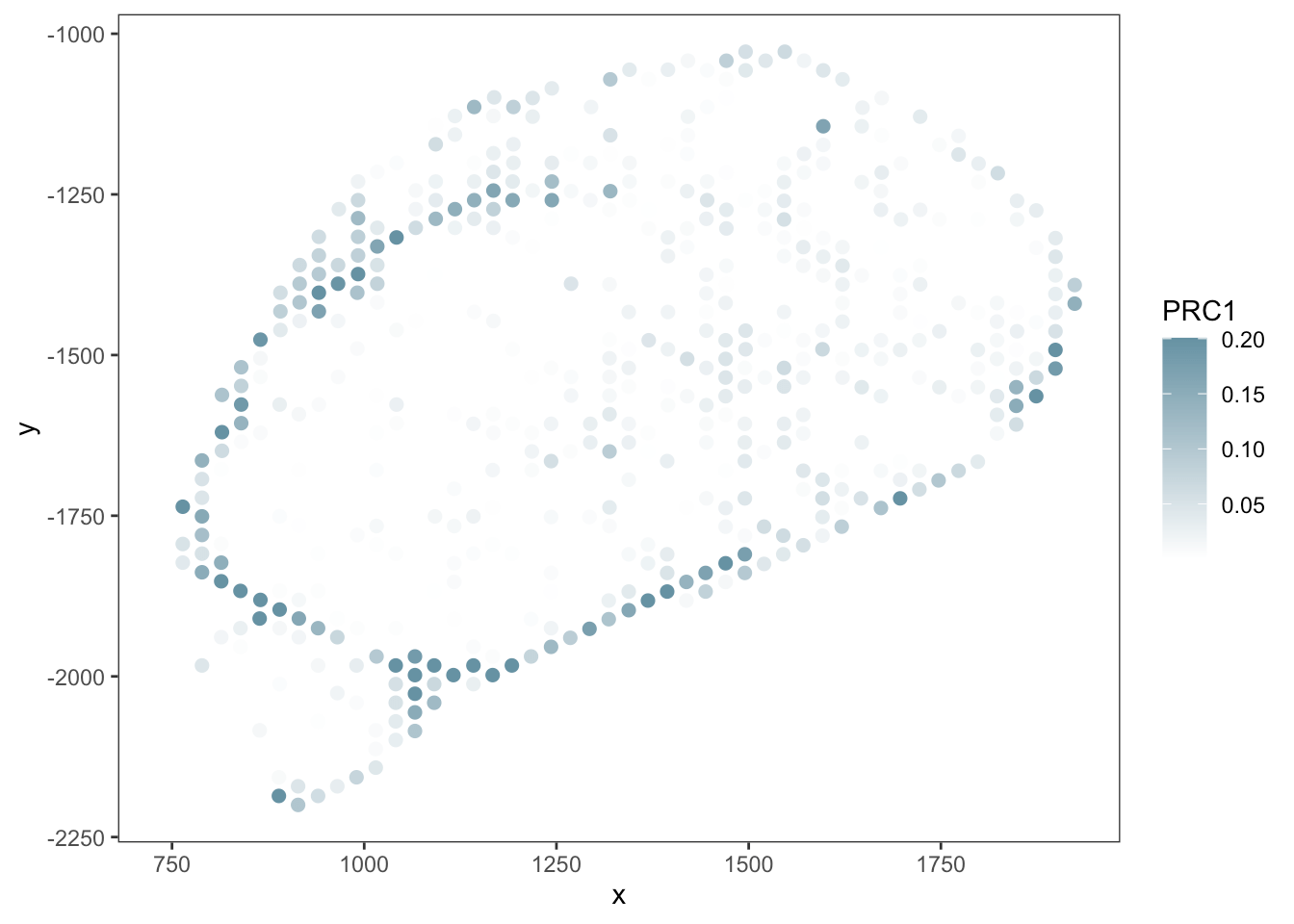
PRC
RCTD
SpaTalk
Downstream analyses - How to decide for a tool?
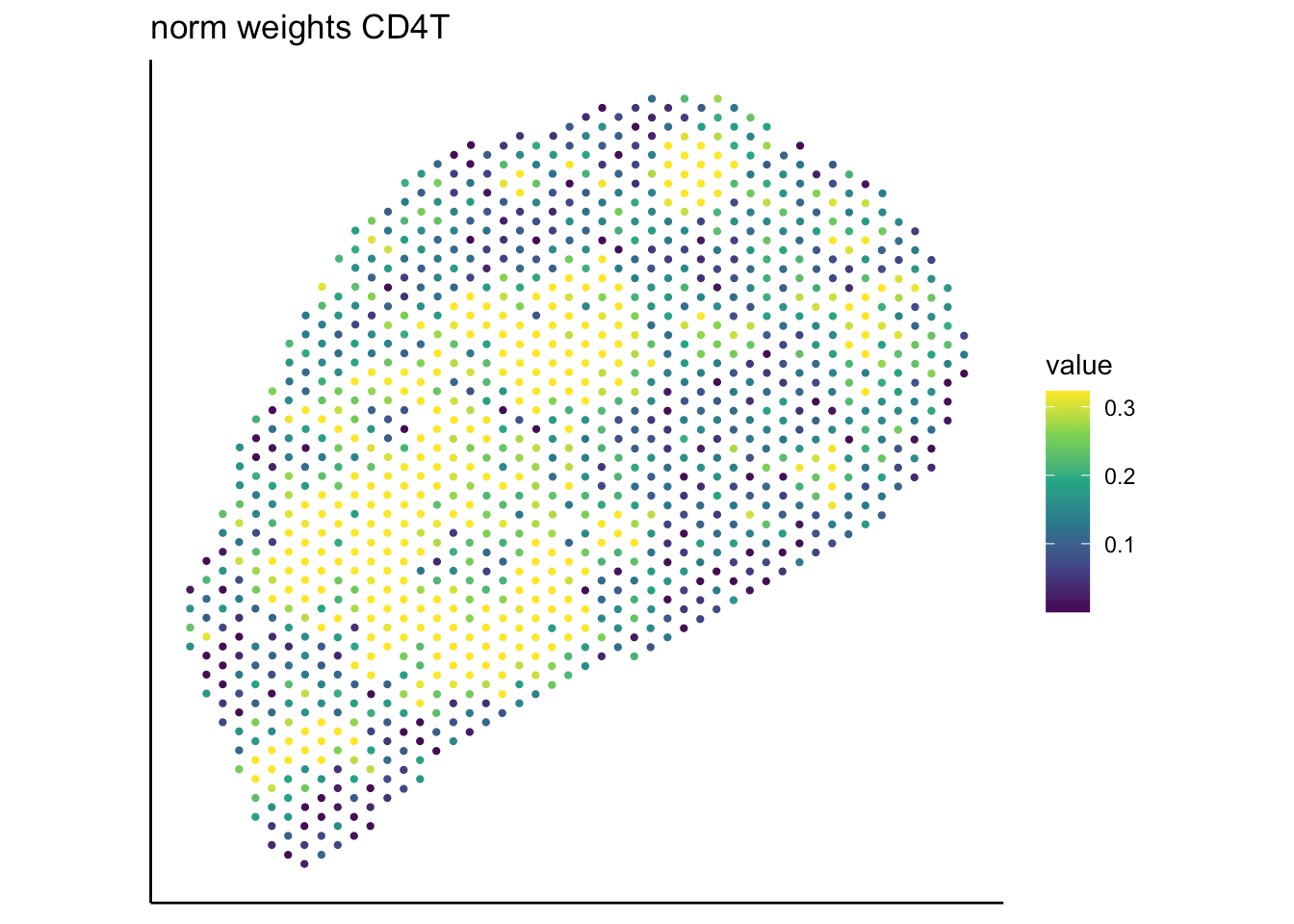
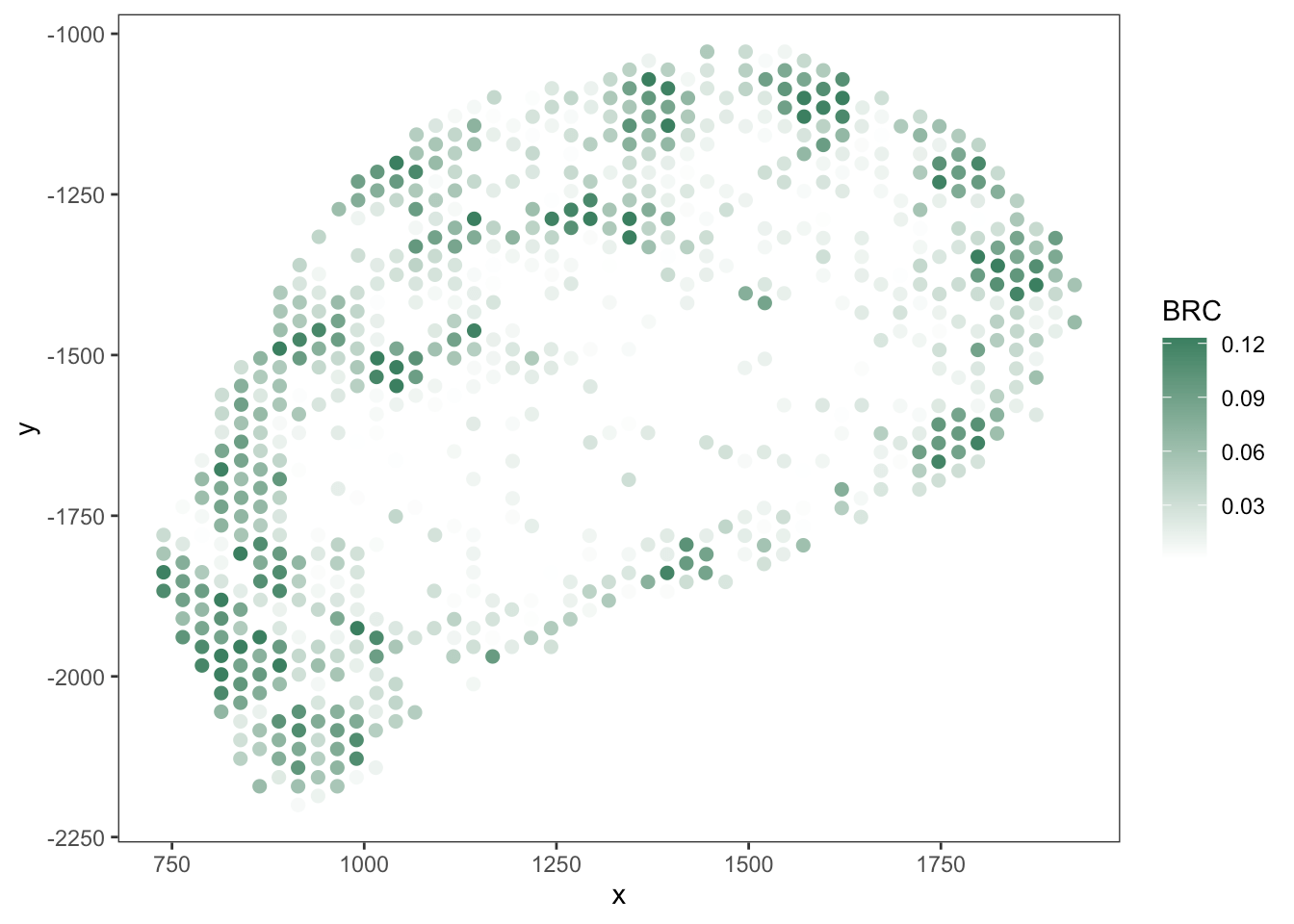
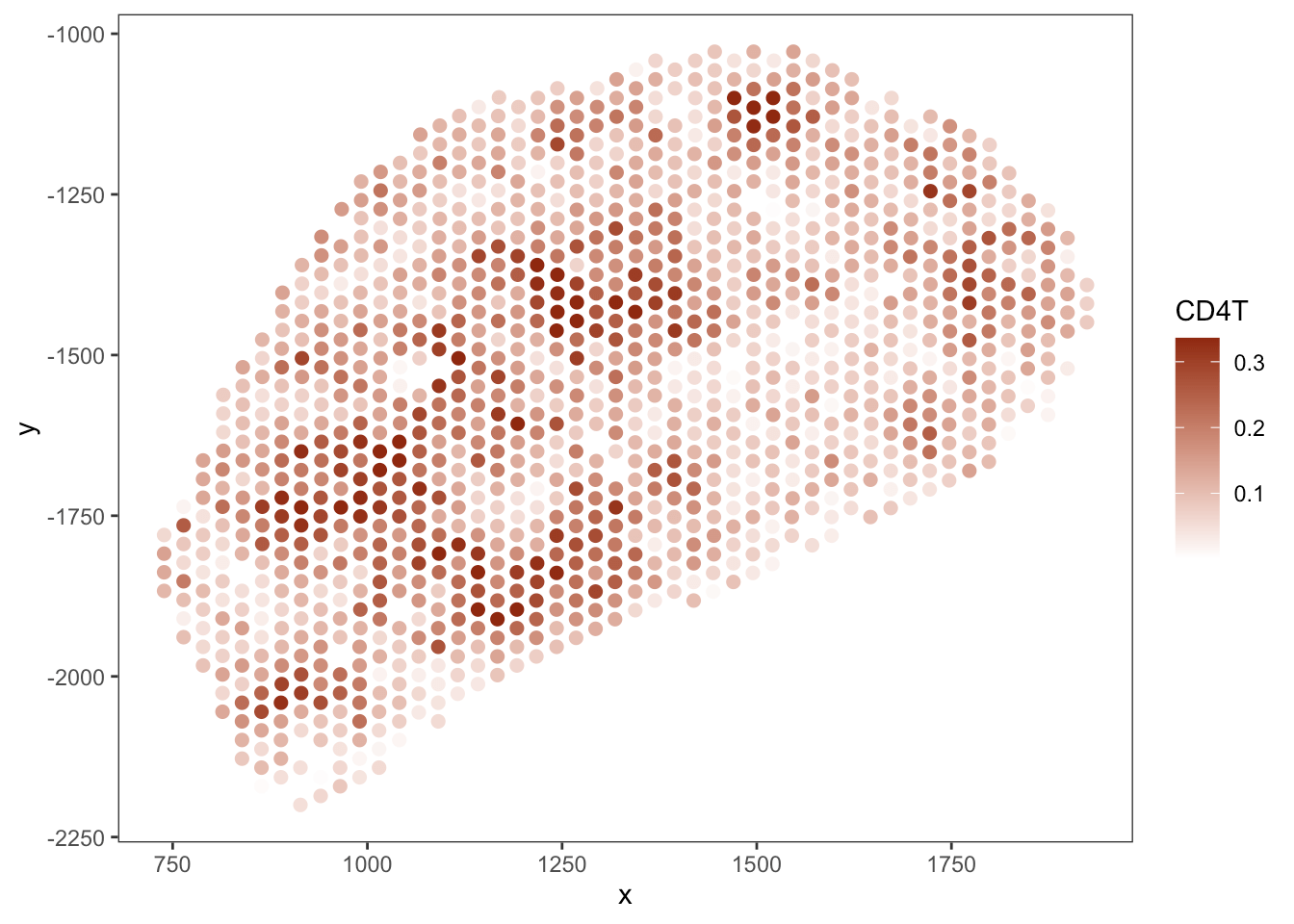
T cell
B cell
RCTD
SpaTalk
PRC
Downstream analyses - How to decide for a tool?
Thank you for your time!
Minimal
By Almut Luetge



