














Jesse Bloom PRO
Scientist studying evolution of proteins and viruses.
Fred Hutch Cancer Center / HHMI
These slides at https://slides.com/jbloom/scripps-circuits-to-systems-2024
Coronaviruses are only RNA viruses with proofreading activity in their polymerase, and so have ~5- to 10-fold lower mutation rate than influenza virus
The average single-nucleotide mutation to SARS-CoV-2 had occurred >10,000 independent times in human-transmitted SARS-CoV-2 by third year of pandemic.
Add some plot here
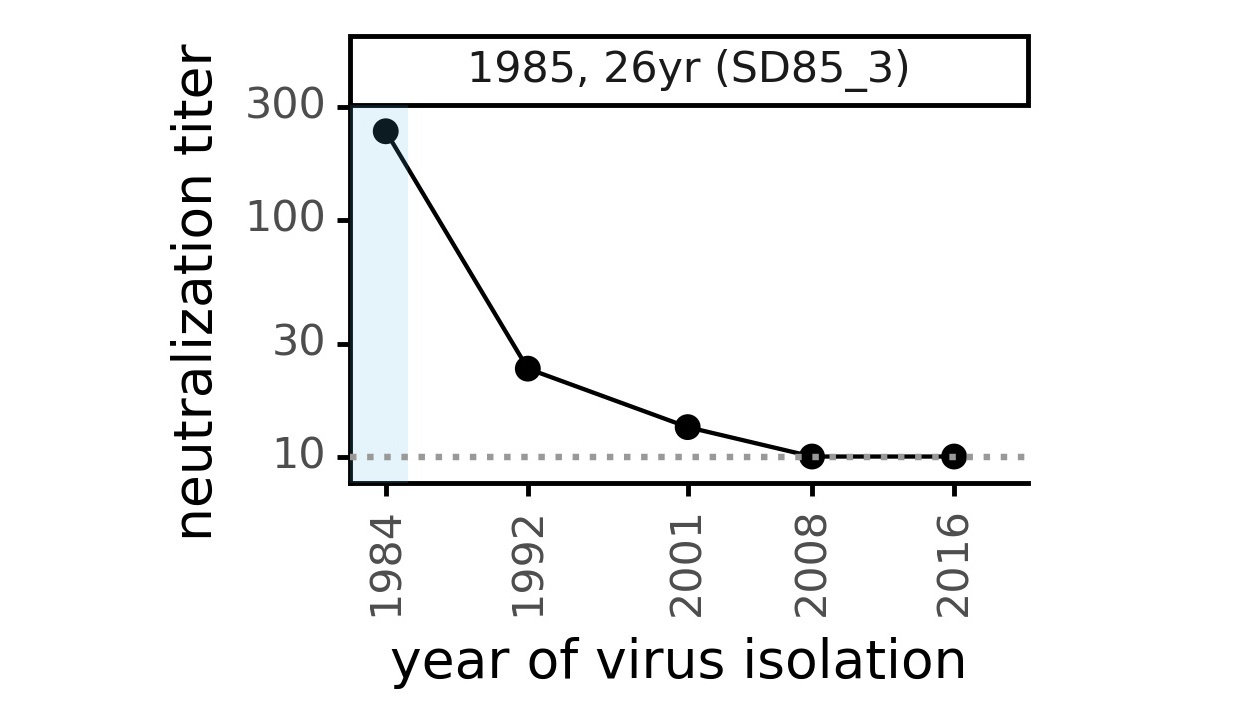
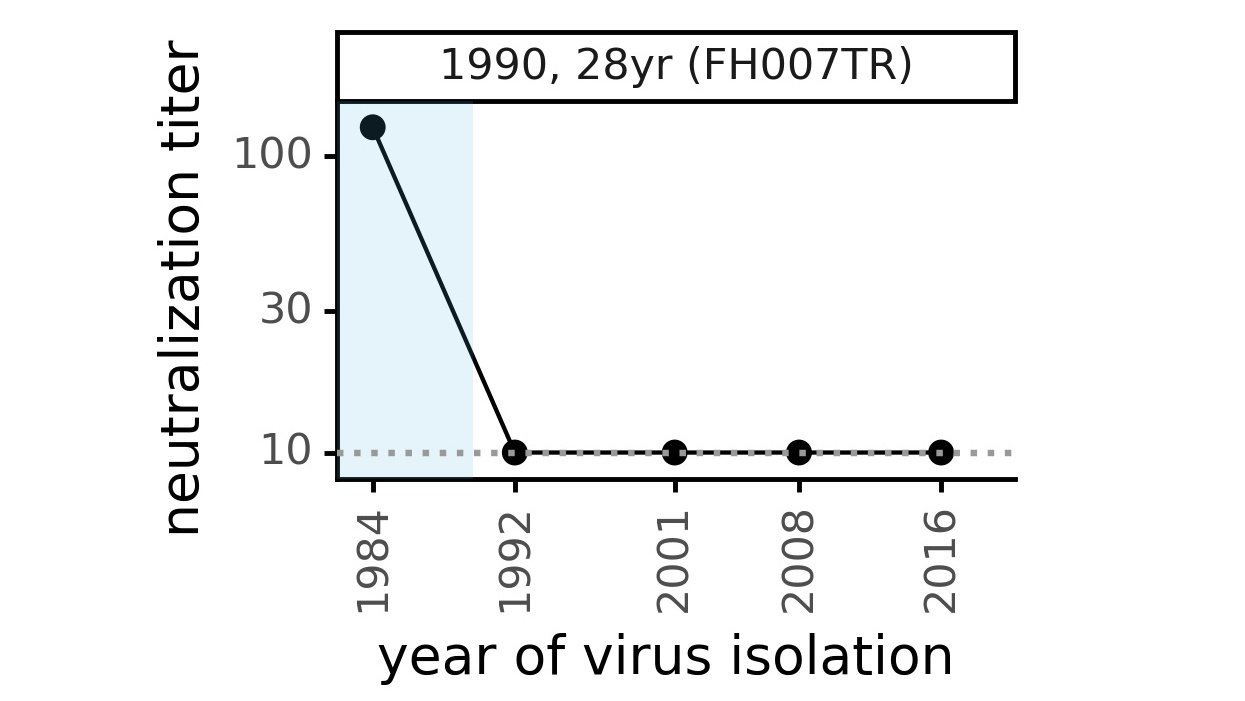
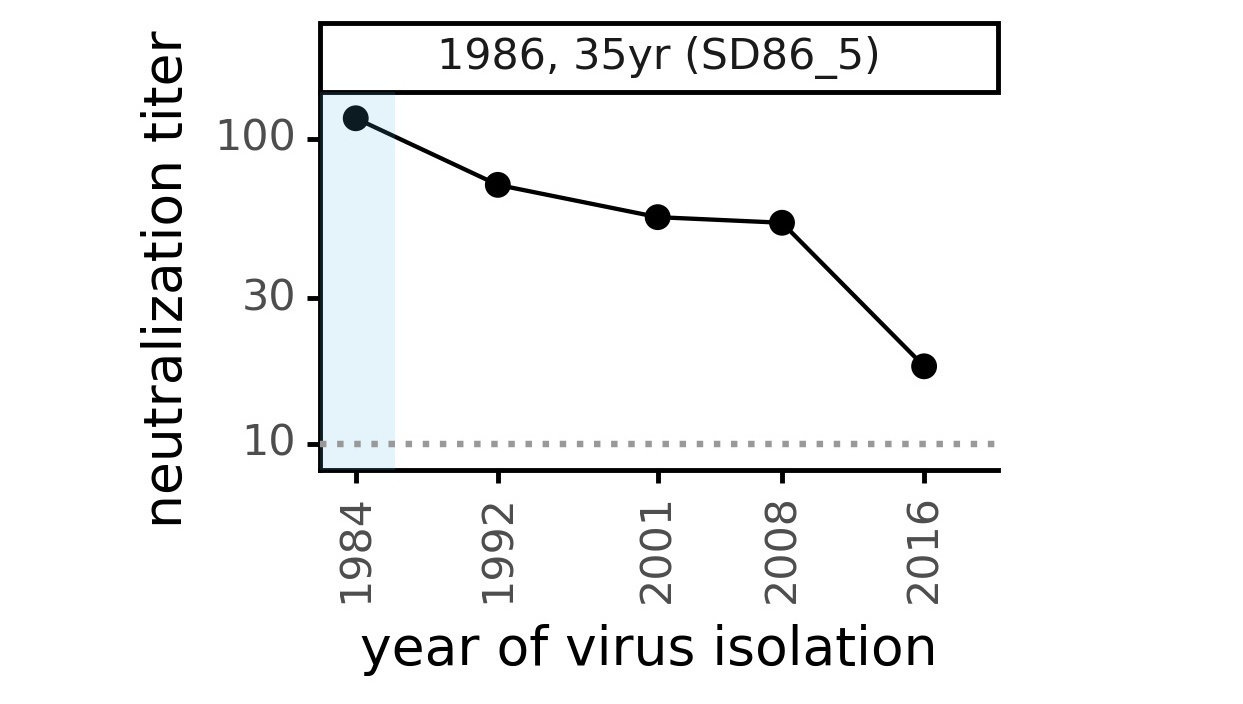
CoV-229E causes common colds and has been circulating in humans for a long time.
Typical person is infected every ~3 to 5 years.
We experimentally generated CoV-229E spikes at ~8 year intervals so we could study them in the lab:
- 1984
- 1992
- 2001
- 2008
- 2016
Sites of evolutionary change in the spike of CoV-229E over the last four decades
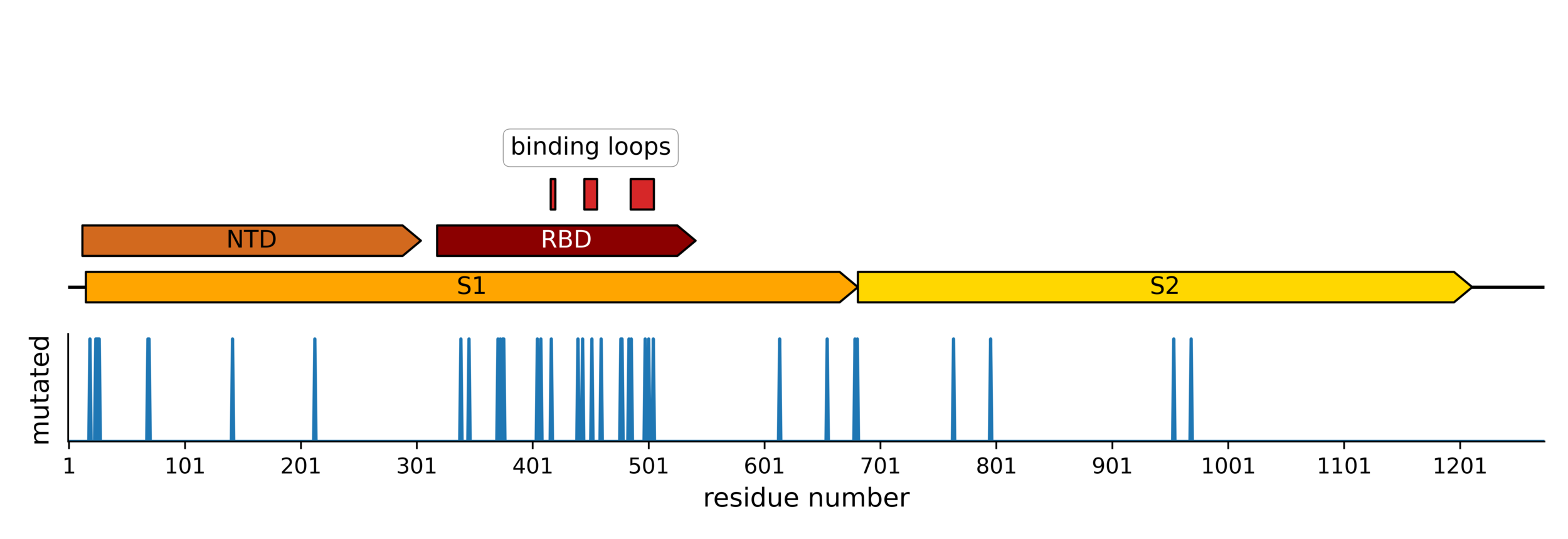
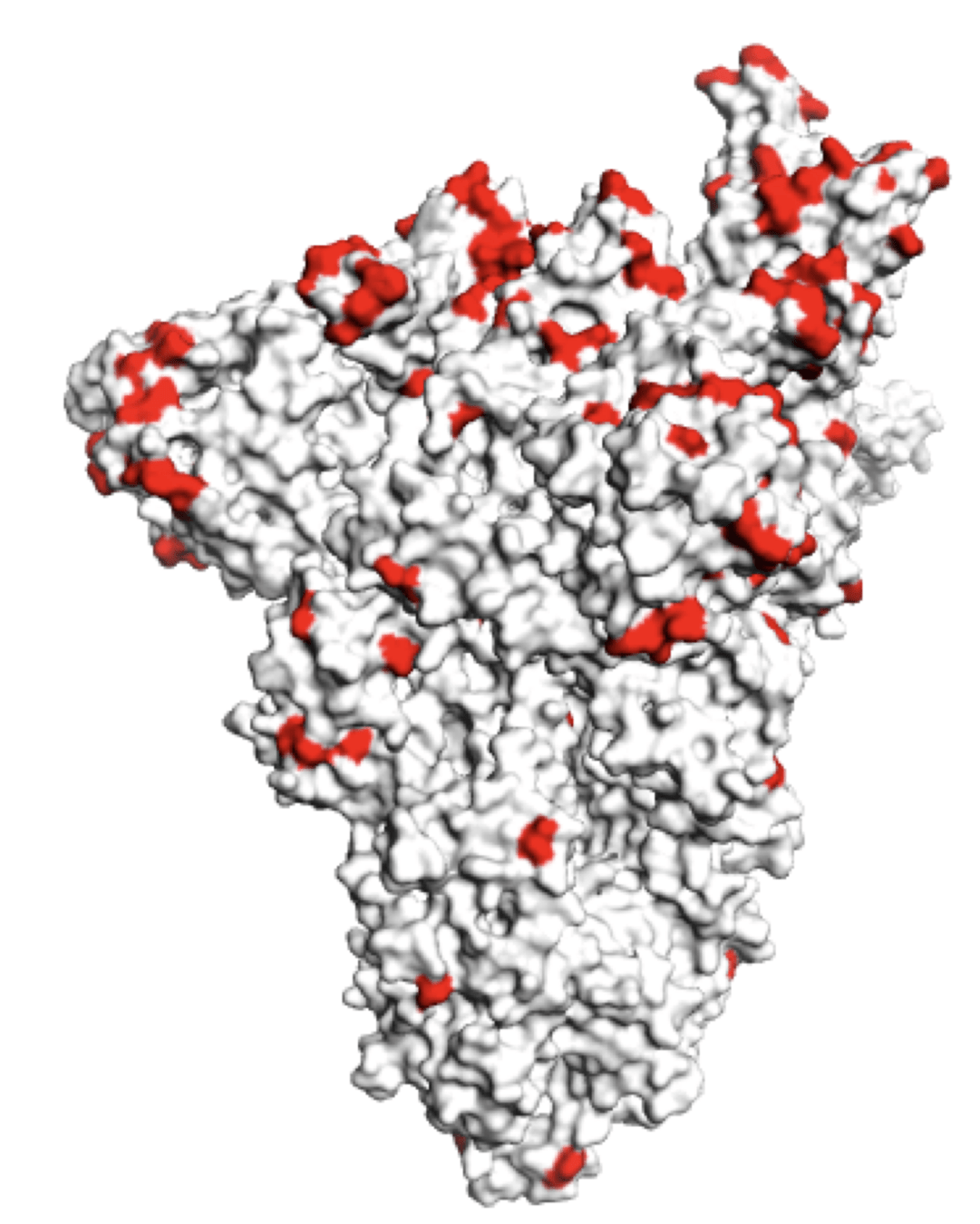
Sites of mutations in SARS-CoV-2 Omicron BQ.1.1 spike relative to Wuhan-Hu-1
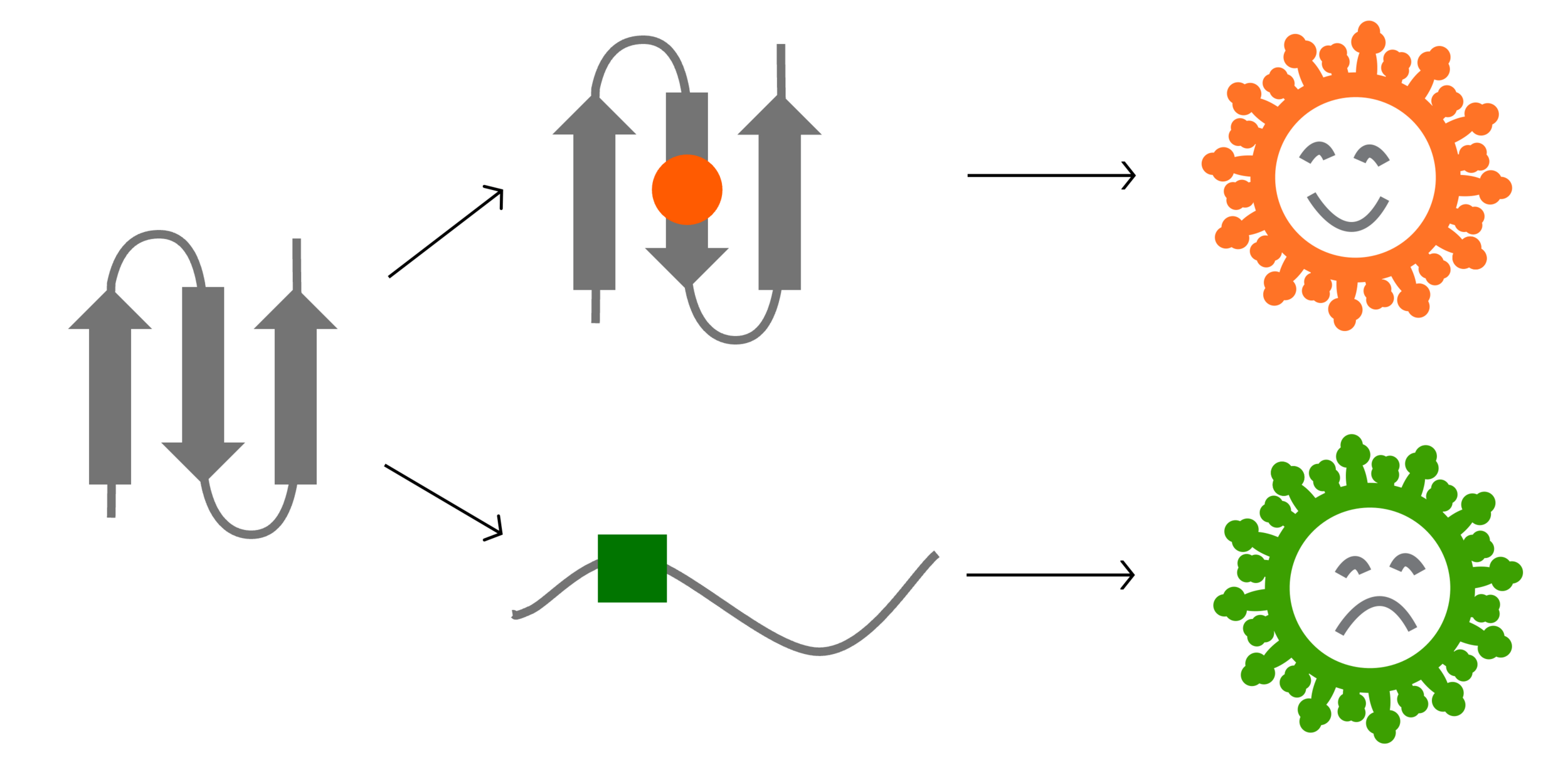
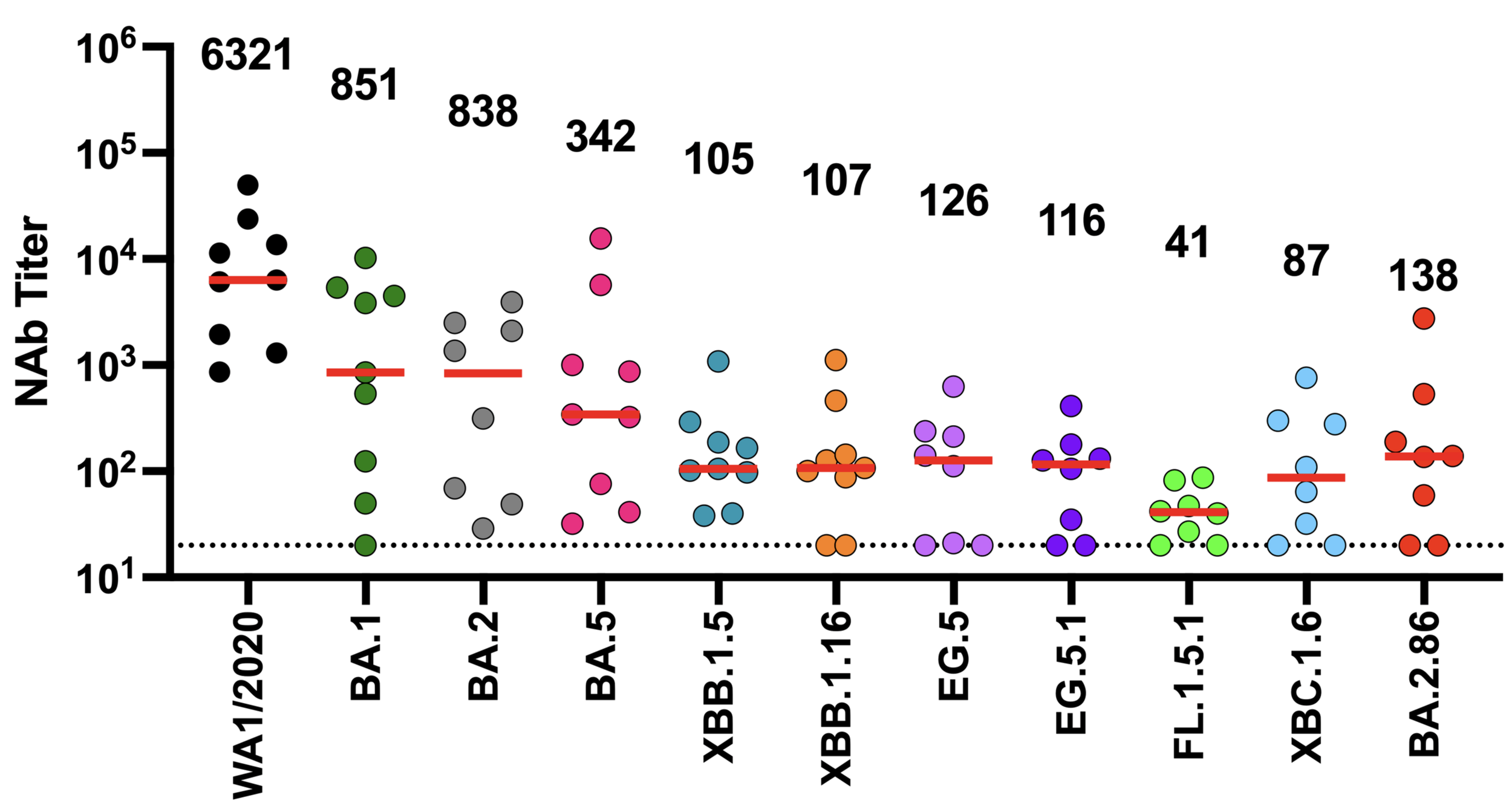
Ideally vaccines would elicit evolution-resistant neutralizing antibodies (like those made by person at right) rather than evolution-sensitive antibodies (like those made by person at left)
neutralization from original COVID-19 vaccine
Original vaccine induced hight neutralizing antibody titers against early viral strains
newer viral variants
neutralization from original COVID-19 vaccine
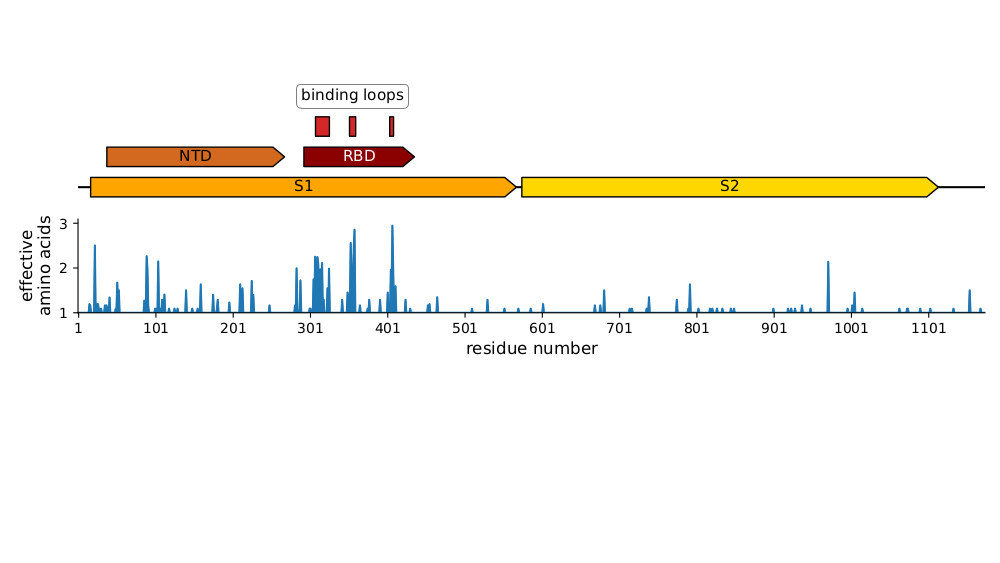
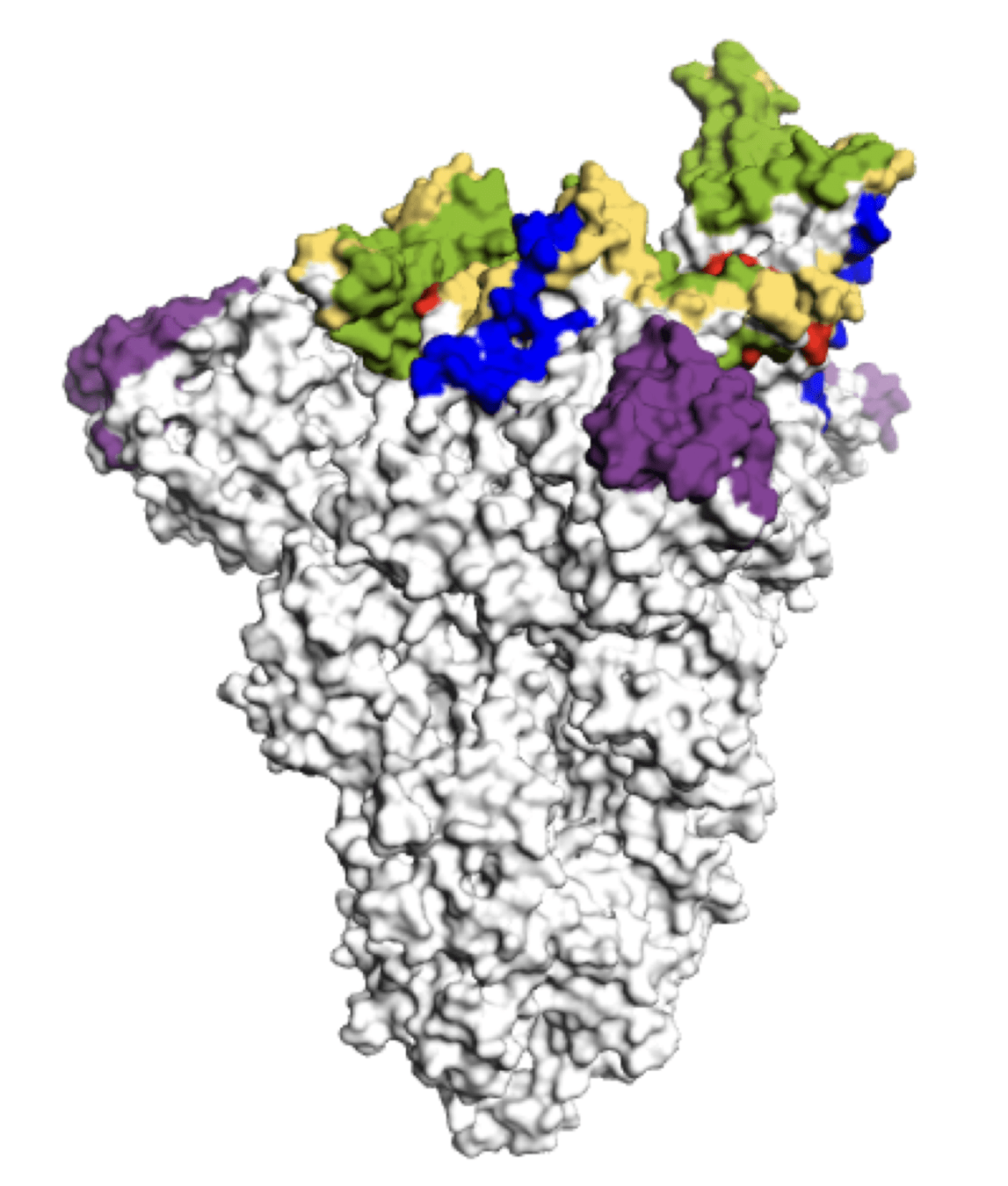
Main regions where neutralizing antibodies bind
Main regions where neutralizing antibodies bind
Sites of mutations in recent (BA.2.86) SARS-CoV-2 strain relative to early 2020 strain
Why only some RNA viruses evolve to escape immunity is a deep question that has not been fully answered (see here for some theories).
But all these viruses have high mutation rates, so explanation has more to do with phenotypic effects of mutations than rate at which they arise.
Rate of viral antigenic evolution
Measles
Mumps
Influenza
SARS-CoV-2
1. Define a virus's evolutionary potential (eg Starr et al 2020; Starr et al 2022)
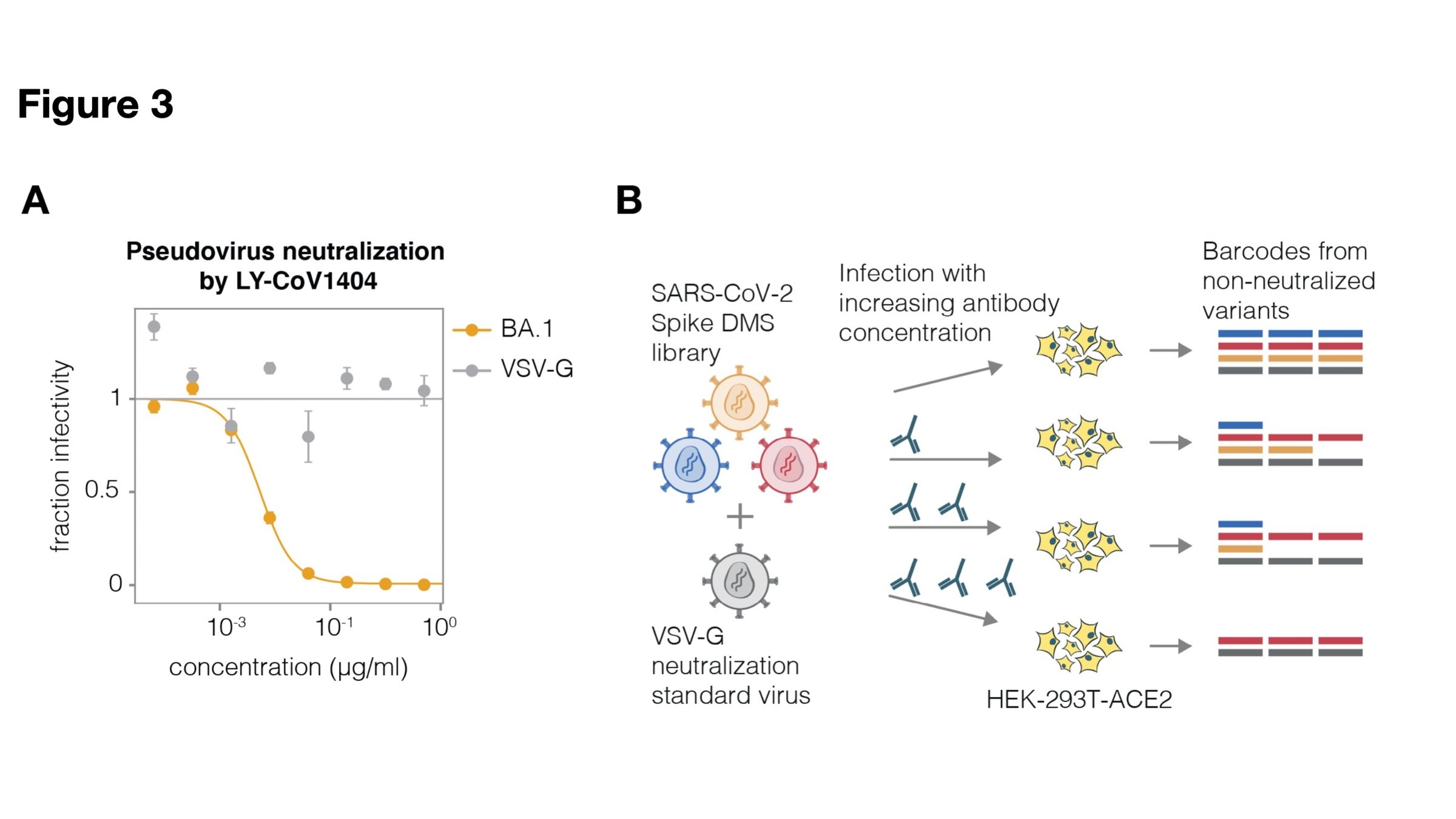
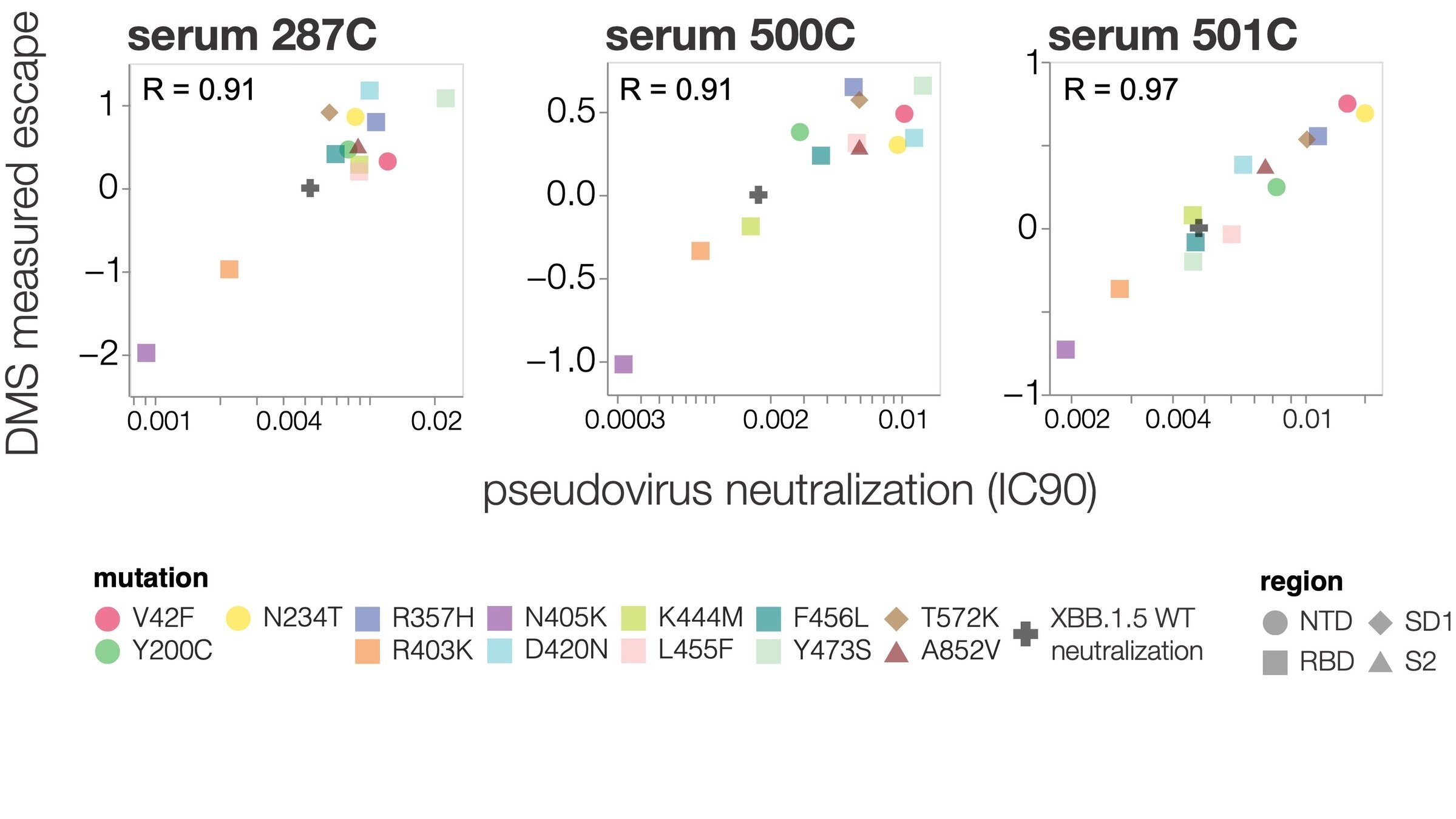
2. Quantify how easily a virus can escape a specific antibody or vaccine (eg Greaney et al 2021; Carr et al 2024)
3. Forecast and interpret viral evolution (eg, Dadonaite et al 2024)
1. Define a virus's evolutionary potential (eg Starr et al 2020; Starr et al 2022)
2. Quantify how easily a virus can escape a specific antibody or vaccine (eg Greaney et al 2021; Carr et al 2024)
3. Forecast and interpret viral evolution (eg, Dadonaite et al 2024)
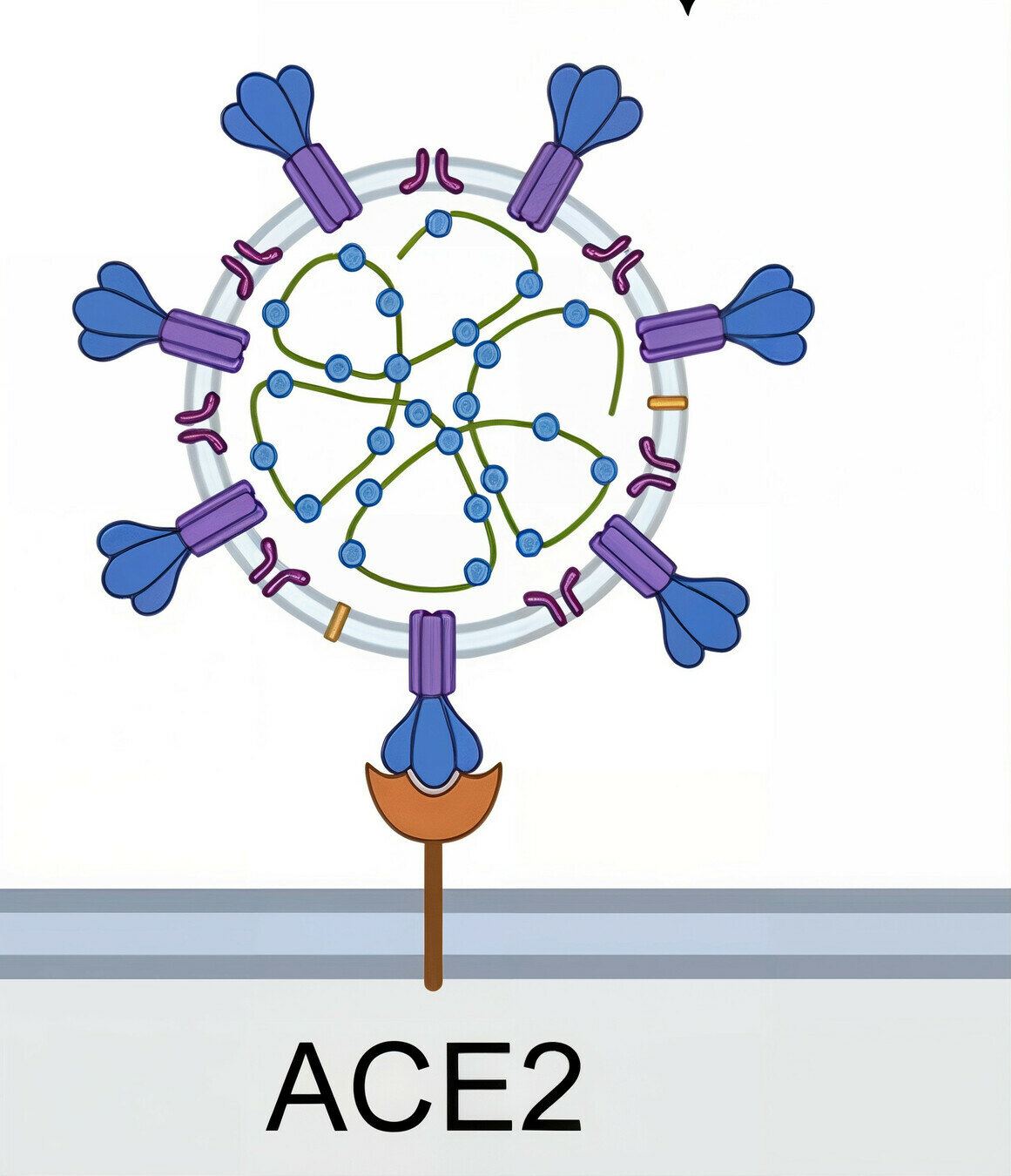
target cell membrane
SARS-CoV-2 virion
spike protein
Image adapted from here
ACE2
viral membrane
cell membrane
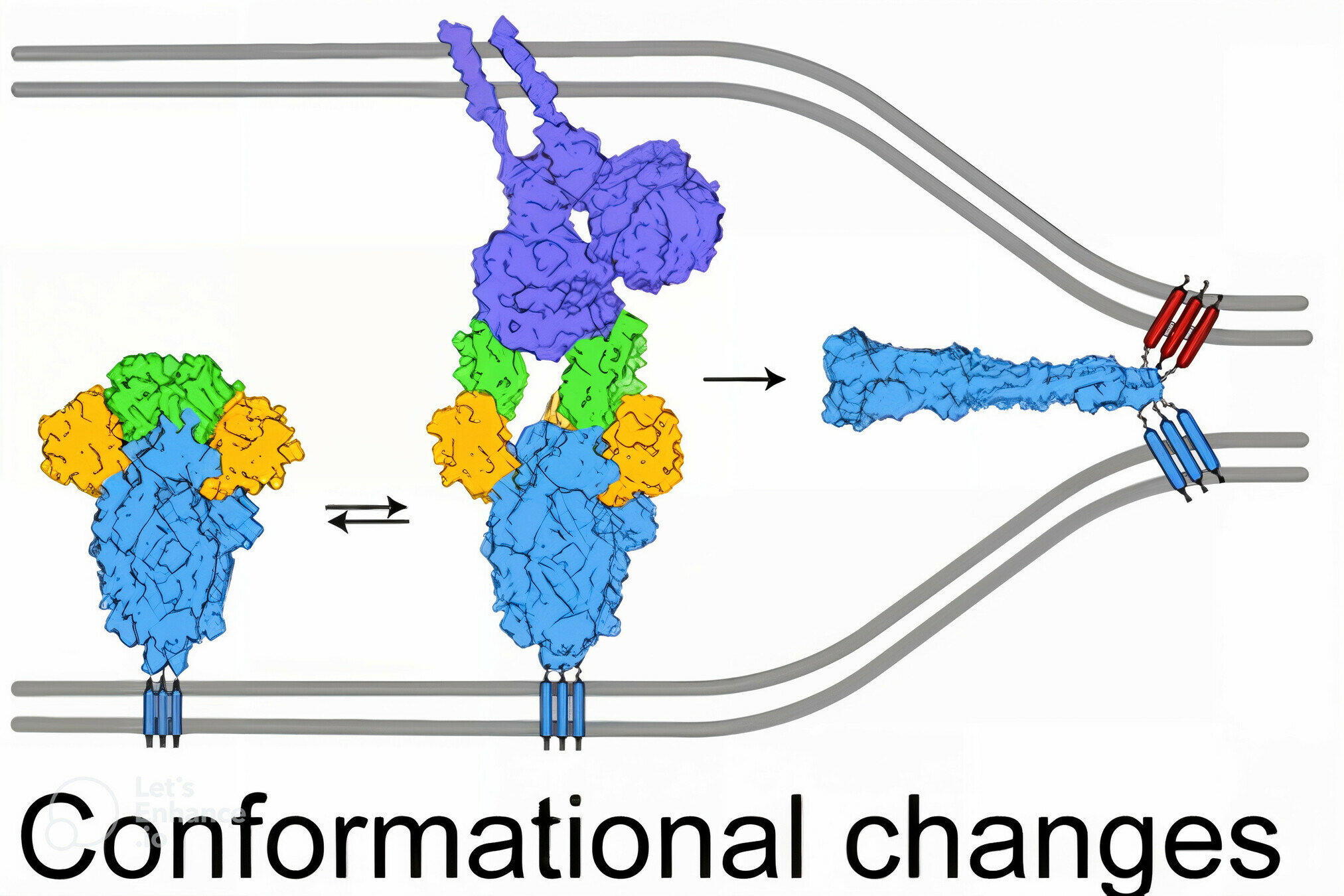
spike
spike conformational change
Image adapted from here
ACE2
antibody
Image adapted from here
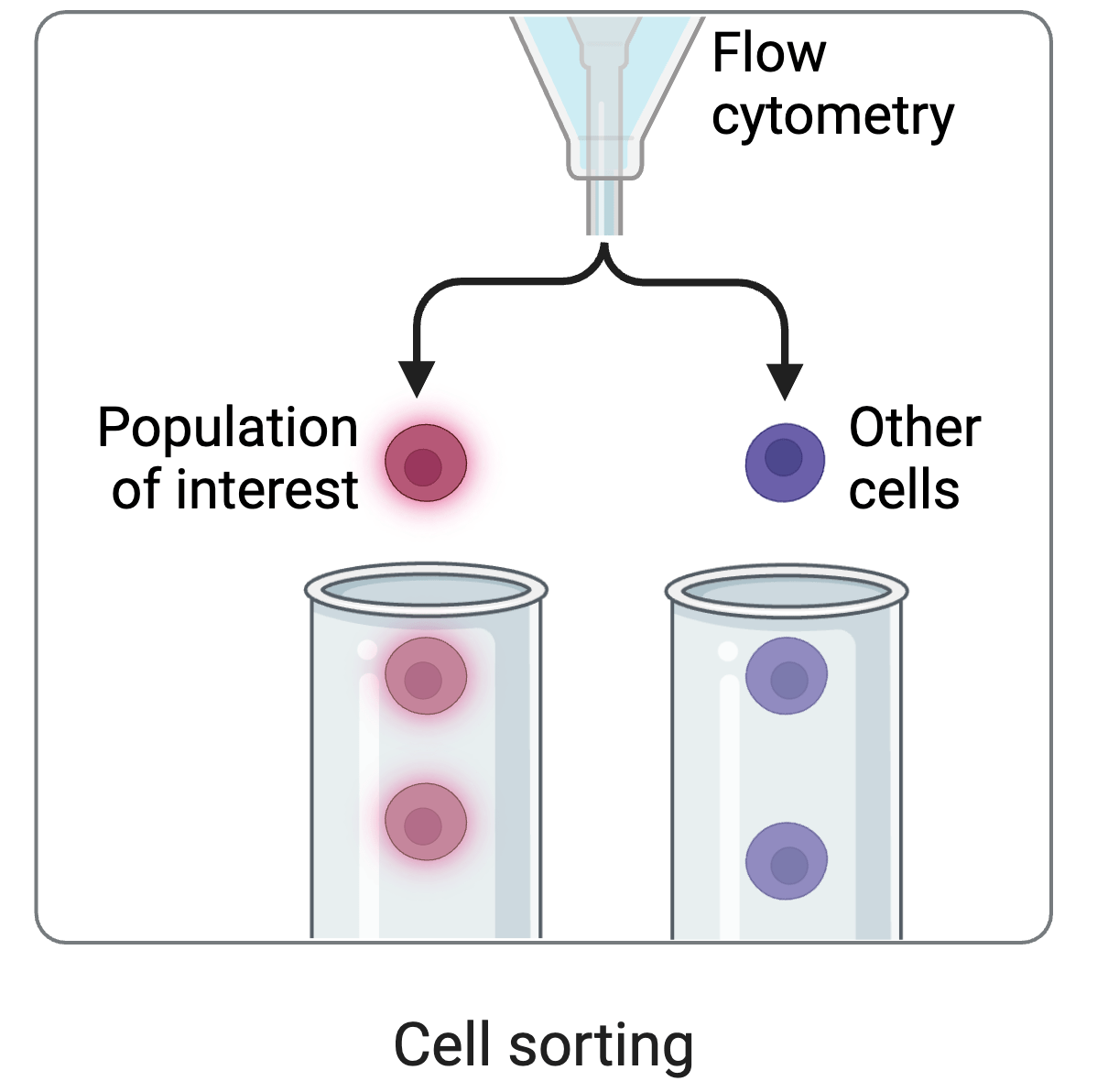
cell sorting
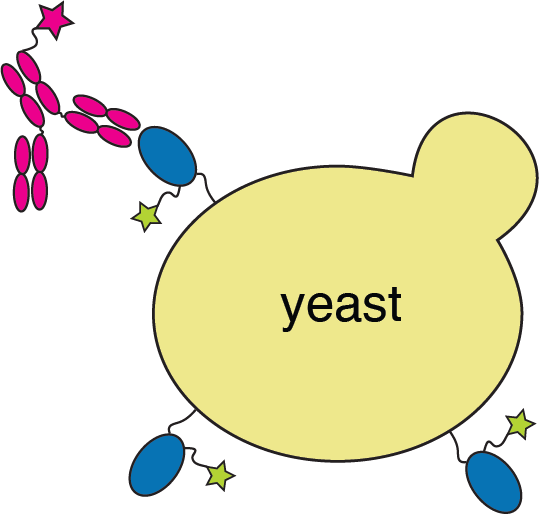
RBD
fluorescent ACE2
yeast
fluorescent tag on RBD
RBD
fluorescently labeled antibody
yeast
fluorescent tag on RBD
See https://jbloomlab.github.io/SARS2-RBD-escape-calc/ for interactive version of this antibody escape calculator (data from our lab and Yunlong Cao's), which has been used >100,000 times
site in RBD
antibody escape
484
417
cell sorting
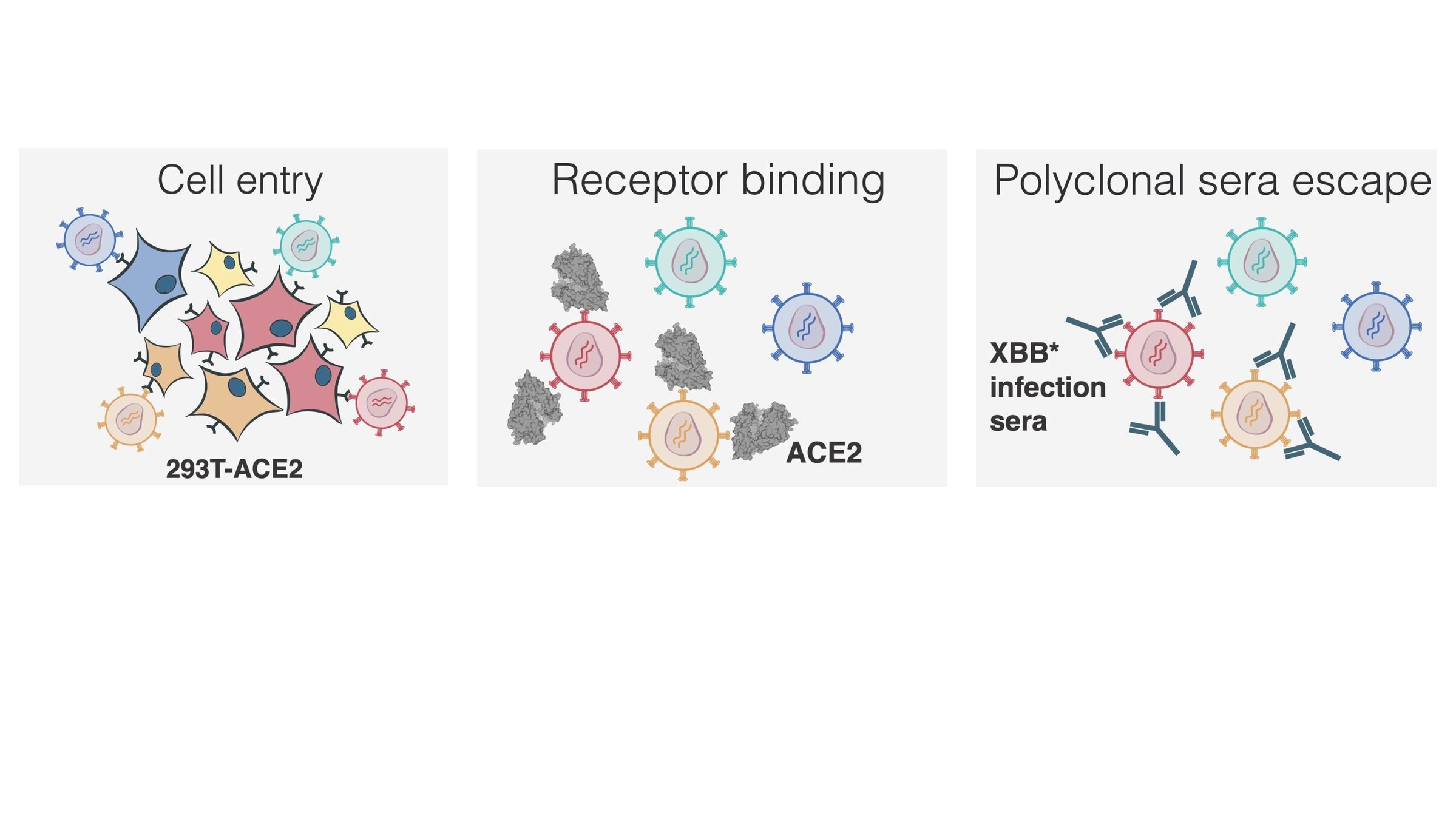
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
actual SARS-CoV-2 virion: pathogen capable of spread in humans
pseudotyped lentiviral particle: not a pathogen, cannot spread in humans
With Trevor Bedford & Ben Murrell
With Trevor Bedford & Ben Murrell
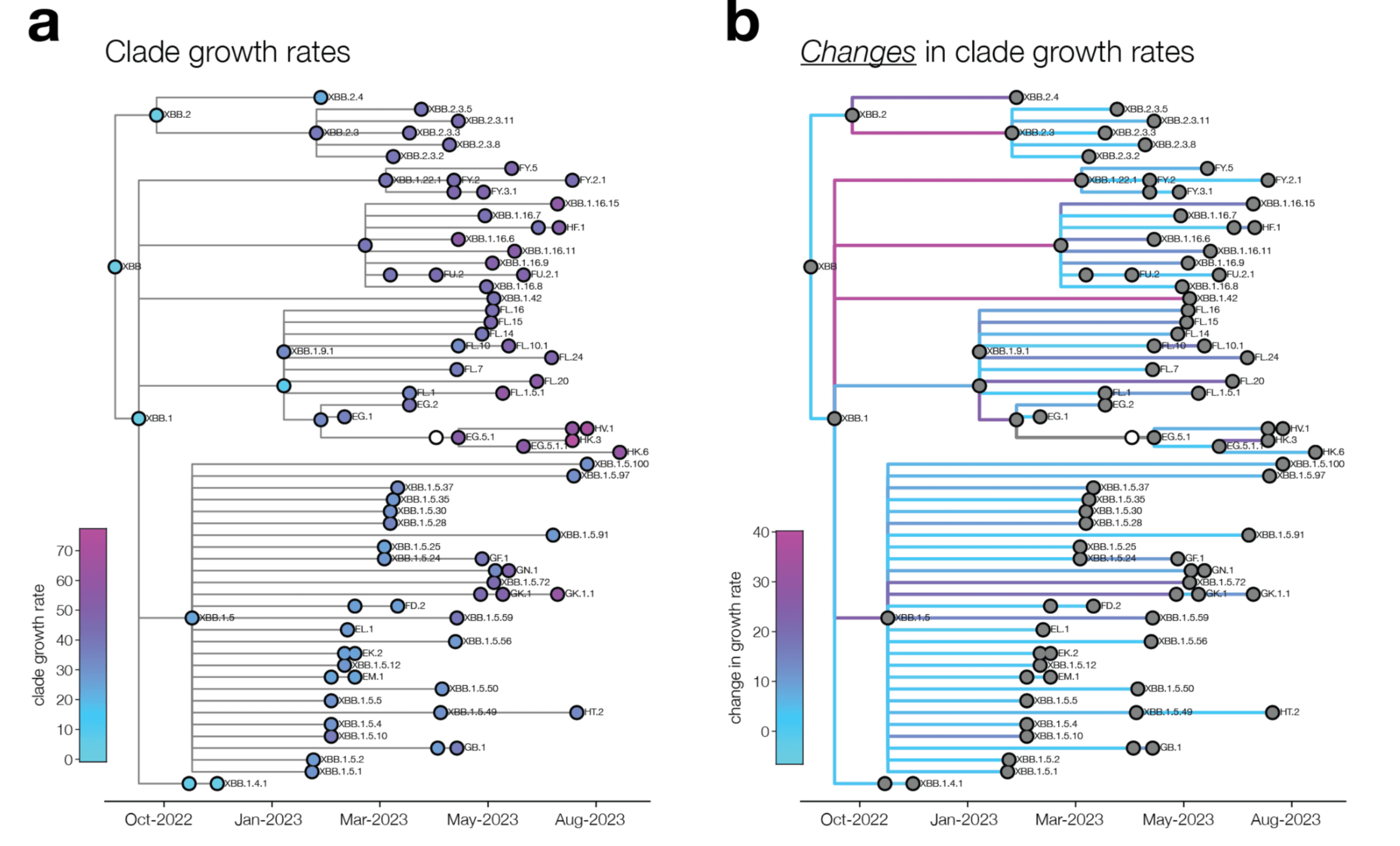
change in clade growth
clade growth
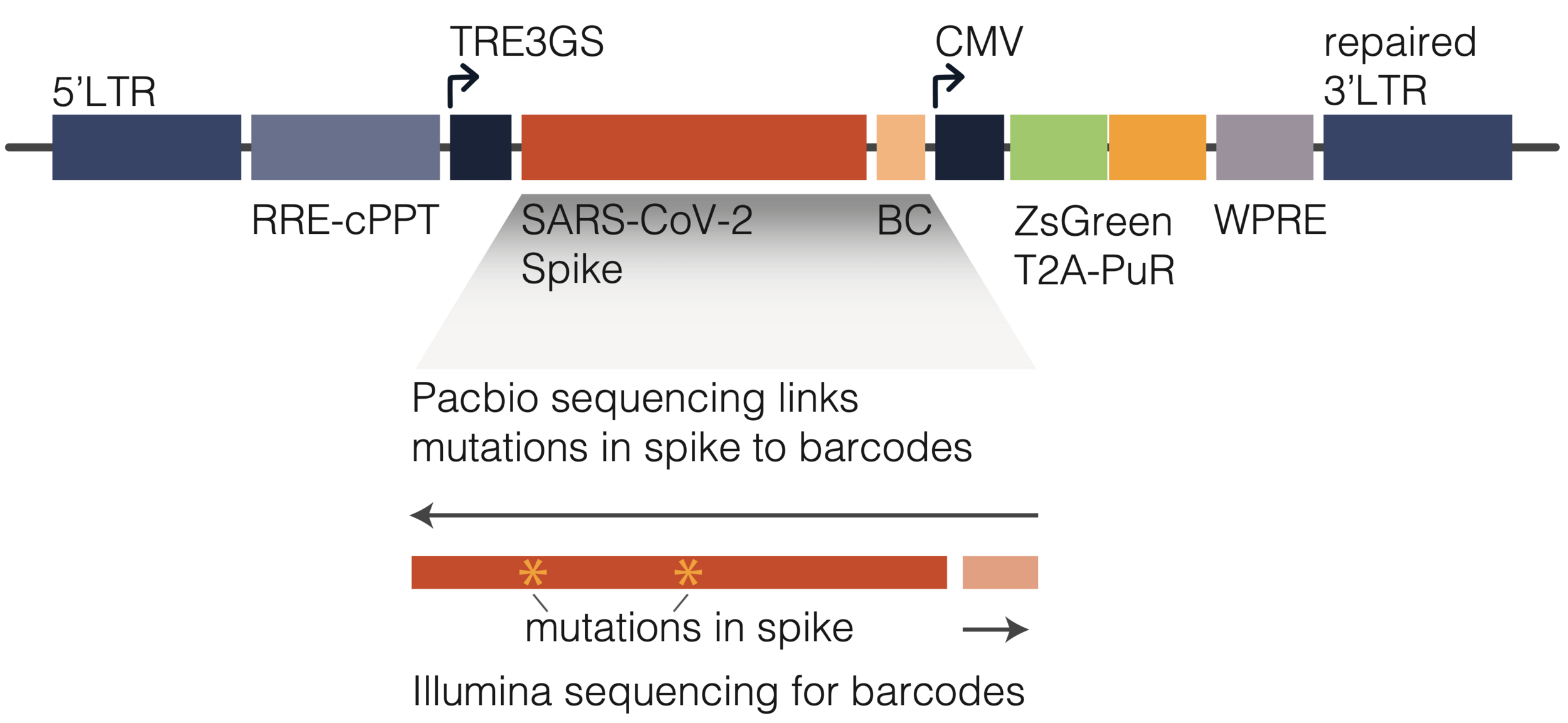
(L122Q, A160T, T199I)
(L122Q, P162Q, T199I)
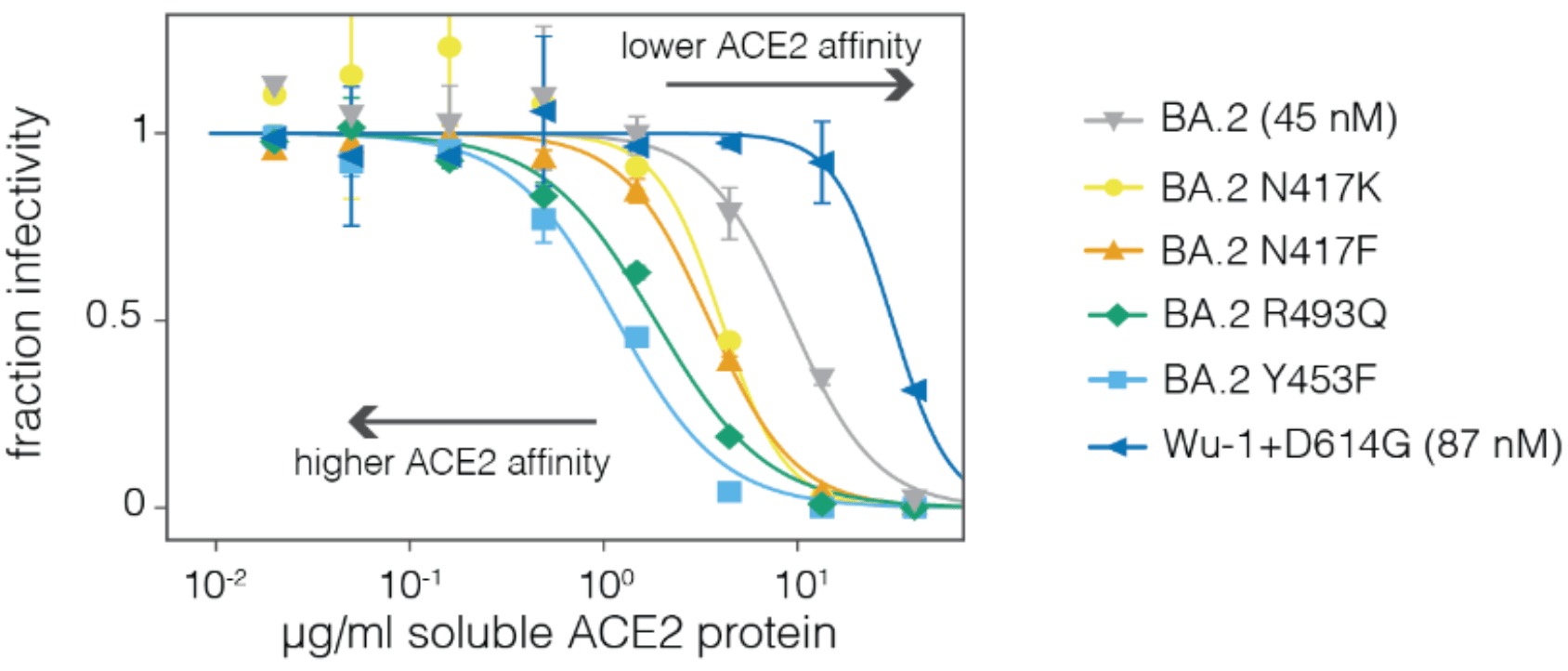
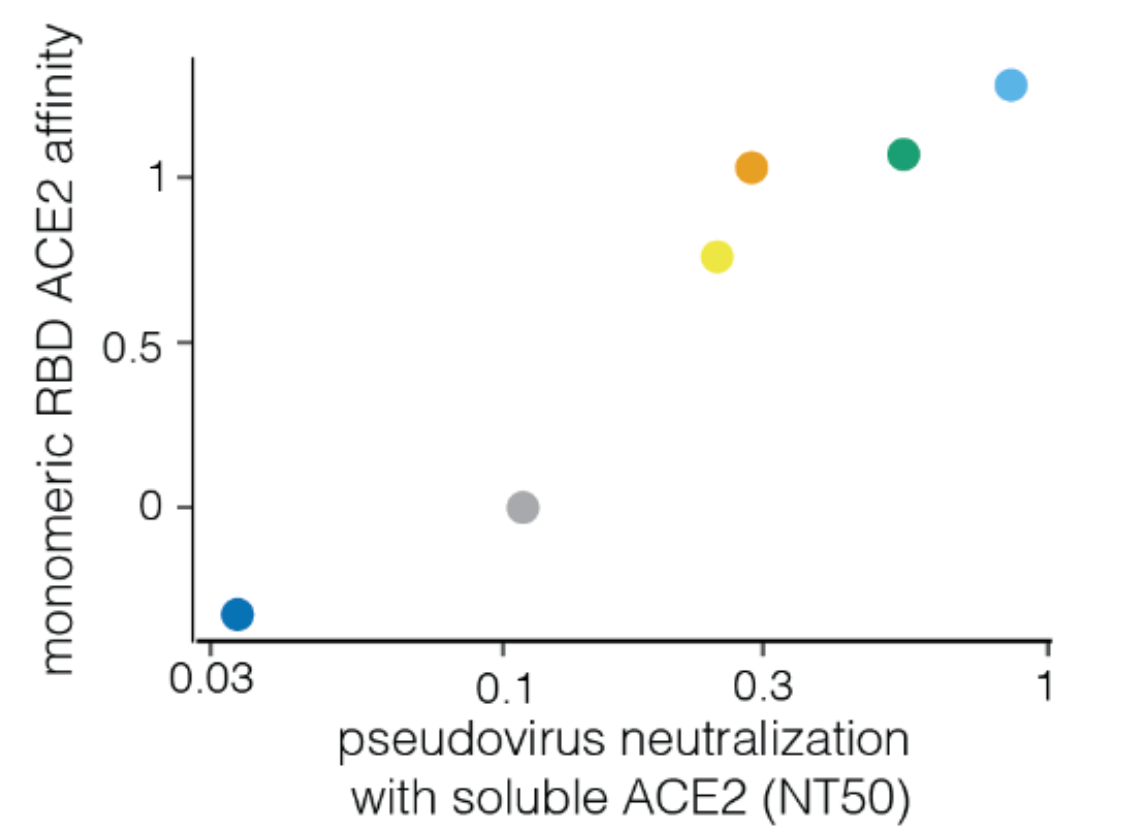
For human endemic (SARS-CoV-2) and potential pandemic (H5N1) viruses, we can safely measure how mutations to entry proteins affect key molecular phenotypes.
For SARS-CoV-2, these measurements can help predict success of variants in humans.
For H5N1, these measurements can help inform surveillance of viral evolution.
Bloom lab
Bernadeta Dadonaite
Kate Crawford
Caelan Radford
Tyler Starr
Allie Greaney
Rachel Eguia
William Hannon
Jenny Ahn
Fred Hutch Cancer Center
Trevor Bedford
John Huddleston
University of Washington
Helen Chu and HAARVI cohort
Neil King
David Veesler
Pirbright Institute
Thomas Peacock
University of Pennsylvania
Scott Hensley
Louise Moncla
Jordan Ort
St Jude Children's Hospital
Richard Webby
By Jesse Bloom
From molecular phenotype to virus evolution: deep mutational scanning of the SARS-CoV-2 spike



